In order to provide the best possible care to patients with a rare condition, it is essential that global knowledge about the condition is gathered. Nationwide, centers of expertise have been set up to stimulate care for rare disorders and to gather knowledge. For the formal recognition of an expertise center by the Ministry of Health, an important condition is that the expertise center gathers, analyzes and shares knowledge through publications. These can be publications in scientific journals, but also treatment guidelines for health care professionals or information brochures for patients or caregivers. We optimize care and research within ENCORE through standardized follow-up and close collaboration between doctors and researchers. That way, we can ultimately develop better treatments for rare conditions. You may therefore be asked to participate in research. Participation in research is always on a voluntary basis. The data obtained is stored and analyzed in an anonymous form. All research has been approved in advance by an ethics review committee.
Genetic testing will be performed on all Angelman Syndrome patients seen in our center of expertise to determine the genetic cause and to be able to support and advise the parents. If genetic testing has already been done elsewhere, it will not be repeated. This genetic knowledge also helps us to better understand the effect of the genetic change ("mutation") on the severity of symptoms. We can then also investigate which treatment works best for a particular mutation. In rare cases, the genetic analysis is inconclusive. In these cases, the genetic change will be further investigated in the laboratory.
The Angelman expertise center is the largest Angelman Syndrome expertise center in the world. We recently published our clinical findings of 100 children and 100 adults with Angelman Syndrome (De Heus, 2019; Besten, 2020).
Detailed knowledge about the course of Angelman Syndrome (which symptoms are present, and when exactly do they arise) is of great importance in order to be able to recognize complaints early and to treat them optimally. We also want to determine how to measure the effectiveness of treatments (‘outcome measures’ research). This is of great importance for drug research (trials). After all, only if we can demonstrate that a new drug is effective, the drug will actually be approved and reimbursed.
Behavioral Treatment for Sleep Problems
Many children with Angelman syndrome have problems falling asleep, staying asleep and wake up early. In our study, we looked whether a behavioral therapy has an effect on the duration of sleep and sleep behavior. Interventions focused on a clear sleep ritual, a fixed sleep rhythm and the minimization of parental visits to the child at night. In addition, we followed a control group for 6 months without giving them any therapy. The results are currently being analyzed and are expected early next year. Experiences and results of the study are currently already being implemented in the outpatient clinic.
Augmentative and alternative communication
In recent years, together with the Dutch AS parent association (vASN), we have worked hard to stimulate augmentative and alternative communication (AAC) in children and adults with Angelman Syndrome. Information from the literature and experiences from practice show that children and adults with AS can learn to communicate through gestures, pictograms and speech computers, but that it is crucial that the environment also communicates with this. In our study, we coach parents and caregivers of people to create a language-rich environment so that children and adults with AS can imitate this language, just as normal children learn to speak by being spoken to. The study has been delayed due to the corona measures, but is expected to be completed by the end of 2020. More information about AAC can be found at www.isaac-nf.nl. To download the Guidance on Supported Communication to which ENCORE has contributed, click here.
The validity and reliability of cognitive tests (‘outcome measures’)
In order to improve treatments for Angelman Syndrome, it is essential that we can determine objectively whether the treatment is effective. Only then can the medicine or treatment be registered and / or reimbursed. Such objective tests are called ‘outcome measures’. Together with the Angelman Syndrome Clinic of Boston and North Carolina (USA), we investigated the validity and reliability of the Bayley III test for use in clinical trials. The Bayley-III is a commonly used developmental test in children with intellectual disabilities. In this study, we analyzed data from more than 100 children, with about half of the children taking the test several times. We investigated whether certain items are relatively easy or difficult for children with AS and whether the test provides a reliable and stable readout over several measurements. This research is relevant for clinical practice and for the upcoming clinical trials. The analysis has now been completed and will be published soon.
3dMD scan
To gain more insight into the effect of the different Angelman Syndrome mutations on brain volume (size of the skull) and facial characteristics, we use a 3dMD scanner. This is a face scanner that uses two pairs of cameras on a large tripod. The cameras create a 180 degree 3D image of the face with real looking skin texture and skin color. During registration, two projectors project random light patterns on the face. The device uses them as a guide for making the image. The process is usually very short, with a maximum of 20 seconds per registration. This means that taking a 3D photo together only takes a few minutes. After scanning, the system automatically generates a 3D image.
The ROSA Study (Rotterdam Outcome Study for children with Angelman syndrome)
For children with Angelman syndrome, there is a lack of suitable measuring instruments (outcome measures). A measuring instrument is a questionnaire, test, or device. What it measures varies by instrument. Standard measuring instruments are often less suitable for children with Angelman syndrome. Problems talking, moving, or concentrating interfere with the results on such a test. Unfortunately, little is known about what are suitable and feasible measuring instruments for children with Angelman syndrome.
Therefore, the aim of this research is to find suitable and feasible measuring instruments for children with Angelman syndrome. These can be used for (medicine) research, and in the future perhaps also in healthcare. A second aim of this research is to collect information about the natural course of Angelman syndrome. This will contribute to our knowledge of this rare and complex disease. Topics we focus on in this study are: brain activity, social functions, nutrition and metabolism, sleep, exercise and walking, and hormone balance.
The ROSA study has now been completed. You can read the results in our publication, click here to open the publication.

Clinical Trials for New Treatments for Angelman Syndrome
Gaboxadol
We have participated in the NEPTUNE study from OVID. In a previous open label study (the STARS trial) a positive effect was seen of gaboxadol on daily functioning in patients with Angelman syndrome (click here for more information). In the ENCORE lab, a positive effect on motor skills in mice with Angelman syndrome was observed. The NEPTUNE studie was an international phase 3 double blind, placebo controlled study. From the Netherlands 4 children participated. Unfortunately, the results showed a similar improvement in children taking gaboxadol and those taking placebo. Click here for more information about the results. The study was recently stopped. These results shows the importance of a placebo controlled trial to ensure good evidence of clinical effectiveness. We thank all the participating families for their help in this study.
Tangelo study
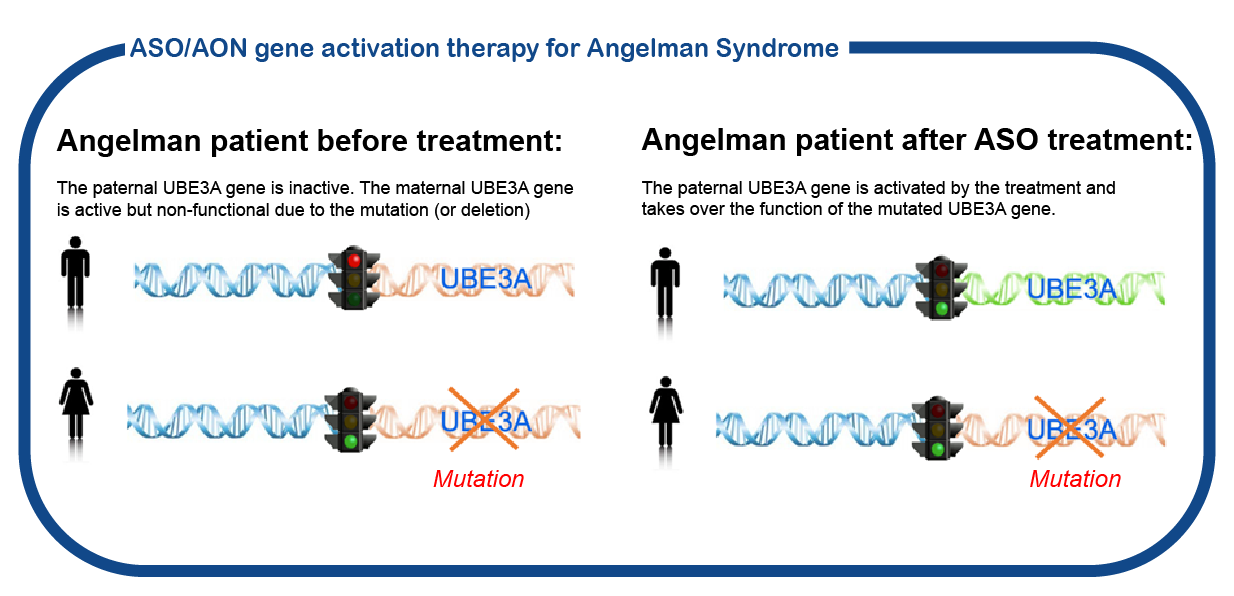
We are currently participating in the Tangelo studie from Roche. This is a large international phase 1/2 study for children between ages 1 and 12 yrs that will receive treatment with an oligonucleotide designed to turn on the paternal allele so that the UBE3A gene is accessible for reading again. Click here for more information about the study. For more information please send an email to angelman@erasmusmc.nl
Together with the vASN We keep parents informed of the latest developments with frequent newsletters and via the website of the parent association. Furthermore, an international AS center partnership has been established to optimize clinical care and to collaborate in scientific research: https://www.angelman.org/angelman-syndrome-clinics/
Halpin S, et al. (2025). Advancing observer-reported outcome measurement: development of the MOOD-AS for observing distress in Angelman syndrome. JPRO, Pubmed
Ten Hooven-Radstaake M, et al. (2025). Criterion Validity, Scalability and Stability of Scoring on the Bayley-III in Children With Angelman Syndrome. JIDR, Pubmed
Loix M, et al. (2025). UBE3A promotes foam cell formation and counters remyelination by targeting ABCA1 for proteasomal degradation. Commun. , Pubmed
Milazzo C, et al. (2025). UBE3A reinstatement restores behaviorand proteome in an Angelman syndrome mouse model of imprinting defects. Autism, Pubmed
Hipp JF, et al. (2025). The UBE3A-ATS antisense oligonucleotide rugonersen in children with Angelman syndrome: a phase 1 trial.Nat med , Pubmed
Navis C, et al. (2025). Language comprehension assessment using the computer-based instrument for low motor language testing (C-BiLLT) in children with Angelman syndrome. Augmentative and alternative communication, Pubmed
van Esbroeck ACM, et al. (2025). Localization of human UBE3A isoform 3 is highly sensitive to amino acid substitutions at p.Met21 position. Mol. Genet., Pubmed
Hagenaar DA, et al. (2025). Age-Related Trajectories of Autistic Traits in Children With Angelman Syndrome. Autism res., Pubmed
Lubbers K, et al. (2024). Autism Spectrum Disorder Symptom Profiles in Fragile X Syndrome, Angelman Syndrome, Tuberous Sclerosis Complex and Neurofibromatosis Type 1. Autism Dev. Disord., Pubmed
Lecoquierre F, et.al. (2024) A recurrent missense variant in the E3 ubiquitin ligase substrate recognition subunit FEM1B causes a rare syndromic neurodevelopmental disorder. Genet Med. Pubmed
Hagenaar DA, et.al. (2024) Outcome measures in Angelman syndrome. J Neurodev Disord. Pubmed
Hagenaar DA, et.al. (2023) Child characteristics associated with child quality of life and parenting stress in Angelman syndrome. J Intellect Disabil Res. Pubmed
Bindels-de Heus KGCB, et.al. (2023) Bone health in children with Angelman syndrome at the ENCORE Expertise Center. Eur J Pediatr. Pubmed
Bindels-de Heus KGCB, et.al. (2023) Hyperphagia, Growth, and Puberty in Children with Angelman Syndrome. J Clin Med. Pubmed
Keary C, et.al. (2023) Gaboxadol in angelman syndrome: A double-blind, parallel-group, randomized placebo-controlled phase 3 study. Eur J Paediatr Neurol. Aug 1;47:6-12. Pubmed
Rotaru DC, et.al. (2023). UBE3A expression during early postnatal brain development is required for proper dorsomedial striatal maturation. JCI Insight. Feb 22;8(4):e166073. Pubmed
Bindels-deHeus KGCB, et.al. (2023) Sleep problems in children with Angelman Syndrome: The effect of a behavioral intervention program. Res Dev Disabil. Feb 6;135:104444. Pubmed
Viho EMG, et.al. (2022) The Hippocampal Response to Acute Corticosterone Elevation Is Altered in a Mouse Model for Angelman Syndrome. Int J Mol Sci. Dec 24;24(1):303. Pubmed
Tanas JK, et.al. (2022) Multidimensional analysis of behavior predicts genotype with high accuracy in a mouse model of Angelman syndrome. Psychiatry. Pubmed
Lubbers K, et.al. (2022) Autism Symptoms in Children and Young Adults With Fragile X Syndrome, Angelman Syndrome, Tuberous Sclerosis Complex, and Neurofibromatosis Type 1: A Cross-Syndrome Comparison. Front Psychiatry. Pubmed
Zampeta FI, Distel B, Elgersma Y, Iping R. (2022) From first report to clinical trials: a bibliometric overview and visualization of the development of Angelman syndrome research. Hum Genet. Pubmed
Pandya NJ, et.al. (2022) A cross-species spatiotemporal proteomic analysis identifies UBE3A-dependent signaling pathways and targets. Mol Psychiatry. Pubmed
Duis J, et.al. (2022) A multidisciplinary approach and consensus statement to establish standards of care for Angelman syndrome. Mol Genet Genomic Med. e1843 Pubmed
Judson MC, el.al. (2021) Dual-isoform hUBE3A gene transfer improves behavioral and seizure outcomes in Angelman syndrome model mice. JCI Insight 6(20):e144712. Pubmed
Pandya NJ, et.al. (2021) Secreted retrovirus-like GAG-domain-containing protein PEG10 is regulated by UBE3A and is involved in Angelman syndrome pathophysiology. Cell Rep Med. 2(8):100360. Pubmed
Milazzo C, Mientjes EJ, et.al. (2021) Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. JCI Insight Aug 9;6(15):145991. Pubmed
Bossuyt S, et.al. (2021) Loss of nuclear UBE3A activity is the predominant cause of Angelman syndrome in individuals carrying UBE3A missense mutations. Hum Mol Genet. Pubmed
Elgersma Y & Sonzogni M. (2021) UBE3A reinstatement as a disease-modifying therapy for Angelman syndrome. Dev Med Child Neurol. Pubmed
Avagliano Trezza T, et.al. (2021) Mono-ubiquitination of Rhabphilin 3A by UBE3A serves a non-degradative function. Sci Rep. 11(1):3007. Pubmed
Den Besten I, et.al. (2020) Clinical aspects of a large group of adults with Angelman syndrome. Am J Med Genet A. Pubmed
Sonzogni M, et.al. (2020) Assessing the requirements of prenatal UBE3A expression for rescue of behavioral phenotypes in a mouse model for Angelman syndrome. Mol Autism. 11(1):70. Pubmed
Geerts-Haages A, et.al. (2020) A novel UBE3A sequence variant identified in eight related individuals with neurodevelopmental delay, results in a phenotype which does not match the clinical criteria of Angelman syndrome. Mol Genet Genomic Med. Pubmed
Zampeta IF, et.al. (2020) Conserved UBE3A subcellular distribution between human and mice is facilitated by non-homologous isoforms. Hum Mol Genet. Pubmed
Bindels-de Heus KGCB, et al. (2020) An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. 182:53–63. Pubmed
Rotaru DC et.al. (2020) Angelman Syndrome: From Mouse Models to Therapy. Neuroscience. 4522:30103-2 . Pubmed
Sonzogni M, et al. (2019) Delayed loss of UBE3A reduces the expression of Angelman syndrome-associated phenotypes. Mol Autism. Pubmed
Tonazzini I, et.al. (2019) The role of ubiquitin ligase E3A in polarized contact guidance and rescue strategies in UBE3A-deficient hippocampal neurons. Mol Autism. Pubmed
Avagliano Trezza R, et al. (2019) Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat Neurosci. 22; 1235–47. Pubmed
Wang T, et.al. (2018) Enhanced transmission at the calyx of held synapse in a mouse model for angelman syndrome. Front Cell Neurosci. Pubmed
Rotaru DC, et.al. (2018) Adult Ube3a gene reinstatement restores the electrophysiological deficits of prefrontal cortex layer 5 neurons in a mouse model of angelman syndrome. J Neurosci. 38; 8011–30. Pubmed
Sonzogni M, et.al. (2018) A behavioral test battery for mouse models of Angelman syndrome: A powerful tool for testing drugs and novel Ube3a mutants. Mol Autism. 14; 9-47. Pubmed
Judson MCC, et al. (2016) GABAergic Neuron-Specific Loss of Ube3a Causes Angelman Syndrome-Like EEG Abnormalities and Enhances Seizure Susceptibility. Neuron. 90; 56–69. Pubmed
Tonazzini I, et.al. (2016) Impaired Neurite Contact Guidance in Ubiquitin Ligase E3a (Ube3a)-Deficient Hippocampal Neurons on Nanostructured Substrates. Adv Healthc Mater 5; 850–62. Pubmed
Elgersma Y. (2015) Neurodevelopmental disease: A molecular tightrope. Nature 526; 50–1. Pubmed
Silva-Santos S, et.al. (2015) Ube3a reinstatement identifies distinct developmental windows in a murine Angelman Syndrome model. J Clin Invest. 125; 2069-76. Pubmed
Steinkellner, T. et al. (2012) Ca(2+)/calmodulin-dependent protein kinase IIα (αCaMKII) controls the activity of the dopamine transporter: implications for Angelman syndrome. J Biol Chem 287, 29627–29635. Pubmed
van Woerden, G.M. et al. (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci 10, 280–282. Pubmed
Elgersma, Y. (2007) Genetic engineering cures mice of neurological deficits: prospects for treating Angelman syndrome. Pharmacogenomics 8, 539–541. Pubmed
van den Ouweland, A.M. et al. (1999) Angelman syndrome: AS phenotype correlated with specific EEG pattern may result in a high detection rate of mutations in the UBE3A gene. J Med Genet 36, 723–724. Pubmed
Fang, P. et al. (1999) The spectrum of mutations in UBE3A causing Angelman syndrome. Hum Mol Genet 8, 129–135. Pubmed
Buiting, K. et al. (1998) Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counseling, and prenatal diagnosis. Am J Hum Genet 63, 170–180. Pubmed
Horsthemke, B. et al. (1996) Familial translocations involving 15q11-q13 can give rise to interstitial deletions causing Prader-Willi or Angelman syndrome. J Med Genet 33, 848–851. Pubmed
van den Ouweland, A.M. et al. (1995) DNA diagnosis of Prader-Willi and Angelman syndromes with the probe PW71 (D15S63). Hum Genet 95, 562–567. Pubmed
Do you have questions about research at ENCORE? Or do you want to participate? Please contact us via encore@erasmusmc.nl or angelman@erasmusmc.nl