Angelman Syndrome (AS) is a severe neurodevelopmental disorder affecting approximately 1:20.000 newborn children. AS mainly affects the central nervous system and causes severe physical and learning disabilities. Children appear entirely normal at birth, but after the first year of life developmental milestones are not met and development entirely stalls at a developmental age of 2 years, regardless of the actual age of the patient. Symptoms may vary between patients, but most commonly include lack of speech, severe intellectual disability, motor deficits, behavioral abnormalities, severely disrupted sleep and seizures. AS patients have a normal life expectancy, but need lifelong daily care.
AS is caused by loss of a functional UBE3A protein. The preclinical research at the ENCORE expertise center focusses on understanding UBE3A function and disfunction, with the ultimate goal to develop potential treatments to ameliorate AS symptoms.
Identifying UBE3A targets.
UBE3A (originally identified as E6-associated protein, E6-AP) is an enzyme with ubiquitin E3 ligase activity. That means it adds ubiquitin molecules to other proteins (called ‘target’ protein). Typically, adding a ubiquitin (Ub) molecule to a target protein will change the activity of the target protein or result in the breakdown of the target protein. An important line of research is to identify the target proteins that are modified by UBE3A. These targets will provide us with important information about the precise role of UBE3A in neuronal function.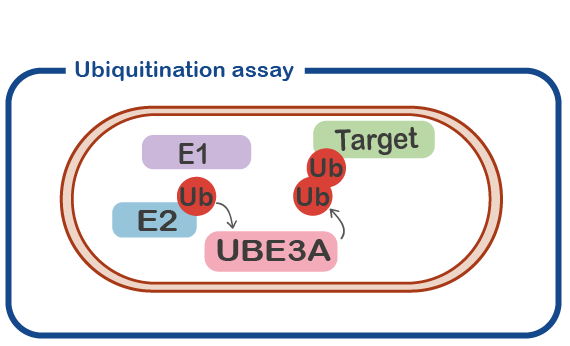
Understanding the effect of UBE3A mutations.
In a subset of patients we find mutations in the UBE3A gene that look quite subtle in which only a single amino acid is replaced by another amino acid (called ‘missense mutation’). This is comparable to a spelling mistake in a word. What is the effect of such a mistake? Sometimes the effect is very severe, and the patients shows a classical Angelman Syndrome phenotype. In other cases, the patient does not look like a typical AS patient, and we are not sure if UBE3A is actually malfunctioning or if a different gene is mutated (See for instance Geerts-Haages, Molecular Genetics and Genome Medicine, 2020). We have now developed assays to measure the effect of missense mutations on the UBE3A protein, by developing a UBE3A ubiquitination assay. This will help us in diagnosing new patients, and it will help us to understand how UBE3A functions.
The role of nuclear UBE3A.
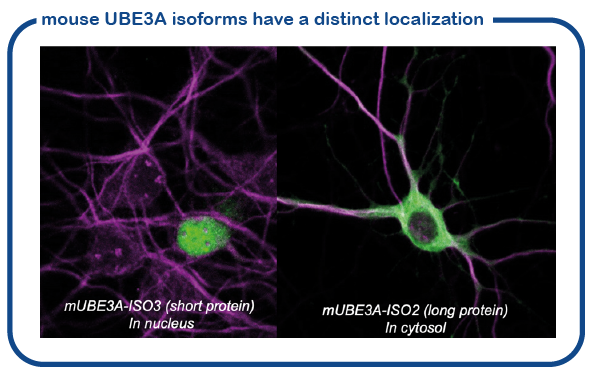
We have recently shown that the mouse makes two slightly different UBE3A proteins (called isoform proteins). These proteins are slightly different in size. The shorter UBE3A protein is specifically localized in the nucleus, the location where the DNA is stored and read. The larger UBE3A protein is present everywhere outside the nucleus (called cytosol). We have recently discovered how this transport to the nucleus is achieved (Avagliano-Trezza, Nature Neuroscience 2019). We further found that in human and mice, most of the UBE3A is located in the nucleus (Zampeta, Human Molecular Genetics 2020), and we have shown that the nuclear UBE3A protein is most important for brain function. A major research line of the lab is now to study the role of UBE3A in the nucleus.
Understanding the role of UBE3A in the brain.
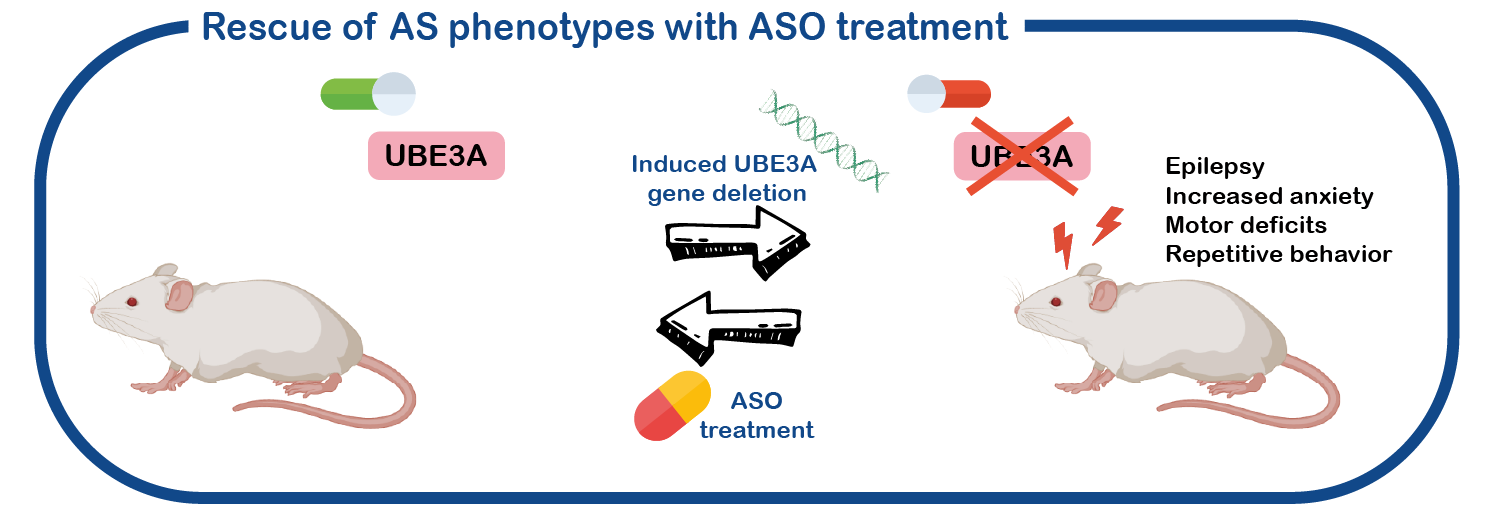
We also study the role of UBE3A in brain function and development. Given that the living human brain is an organ that cannot easily be studied, we use a mouse model for AS that has several symptoms as observed in AS patients (epilepsy, motor deficits, increased anxiety, repetitive behavior). Like the patients, this mouse is lacking a functional UBE3A gene.
An additional advantage of our mouse model system lays in a genetic trick, such that we can switch the UBE3A gene on or off at any given time. We can also switch the gene on or off in specific parts of the brain (Silva-Santos, J. Clinical Investigation, 2015; Sonzogni, Molecular Autism 2019, 2020). This enables us to study its role in brain development, and also determine which brain area is mostly affected. Using electrophysiology, the produced electrical signals in the brain can be assessed and compared between AS and control mice and can be correlated to the behavior of these mice. We are currently focusing on the role of UBE3A in brain development and on the role of UBE3A in the striatum, a specific part of the brain that is possibly responsible for motor and behavioral phenotypes.
Towards a treatment for AS.
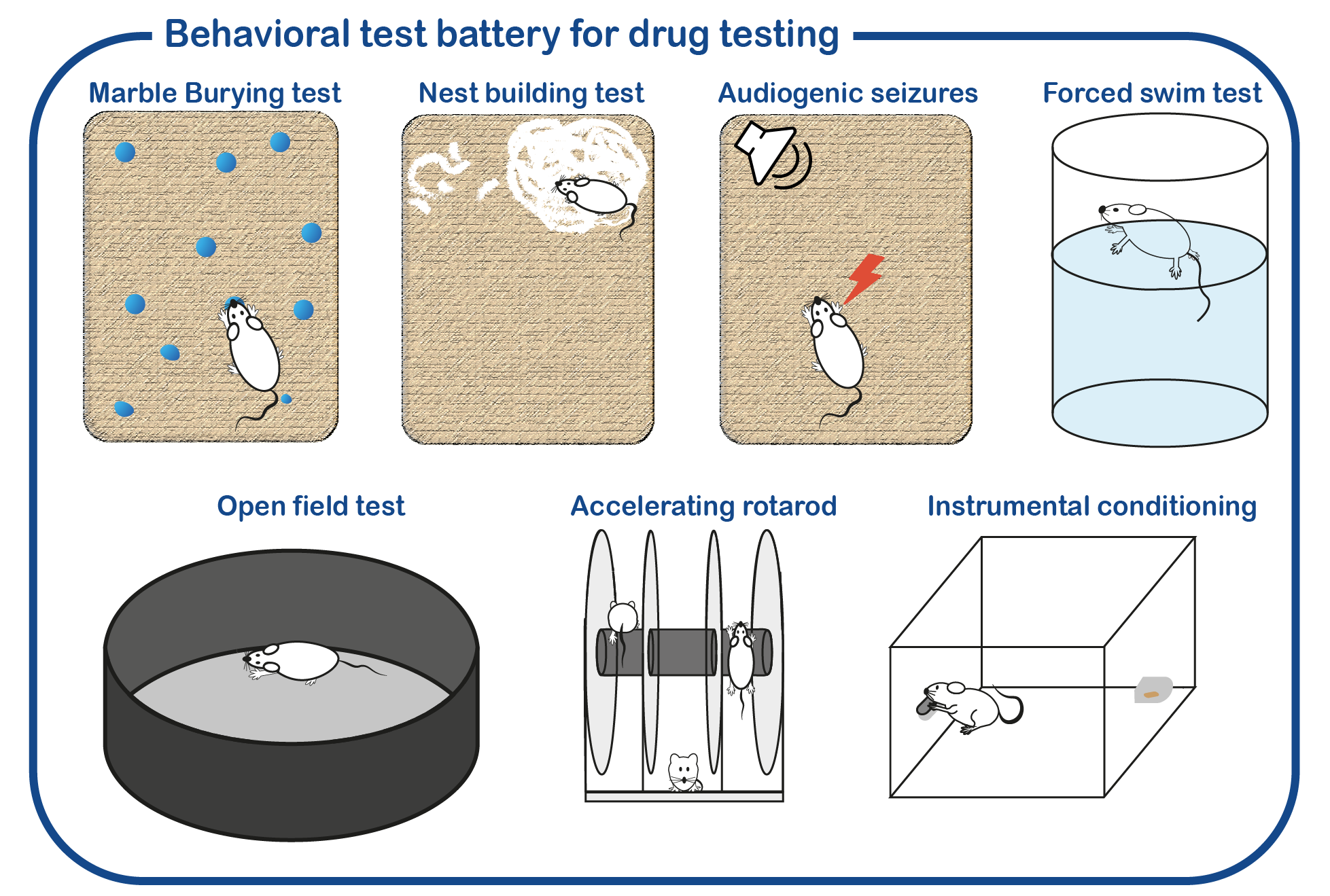
In addition to fundamental studies on UBE3A and its relation to AS, we are also investigating potential therapeutic approaches to alleviate the symptoms of AS patients. To that purpose, we have developed a standardized screening protocol which we use to assess the effect of drugs on AS mouse behavior (Sonzogni, Molecular Autism, 2018). We are continuously testing novel drugs that can potentially correct neuronal function. Besides conventional (small molecule) therapies, we are also employing genetic strategies to restore UBE3A function. This includes the use of antisense oligonucleotides (ASOs) to activate the (paternal) UBE3A gene. ASOs are small pieces of DNA/RNA that can bind RNA molecules. In the case of Angelman Syndrome, these ASOs can restore the synthesis of UBE3A, by targeting the Ube3a-ATS RNA which is responsible of repressing the paternal UBE3A gene. Breakdown of the Ube3a-ATS RNA would restore UBE3A expression in AS patients.
Besides conventional (small molecule) therapies, we are also employing genetic strategies to restore UBE3A function. This includes the use of antisense oligonucleotides (ASOs) to activate the (paternal) UBE3A gene. ASOs are small pieces of DNA/RNA that can bind RNA molecules. In the case of Angelman Syndrome, these ASOs can restore the synthesis of UBE3A, by targeting the Ube3a-ATS RNA which is responsible of repressing the paternal UBE3A gene. Breakdown of the Ube3a-ATS RNA would restore UBE3A expression in AS patients.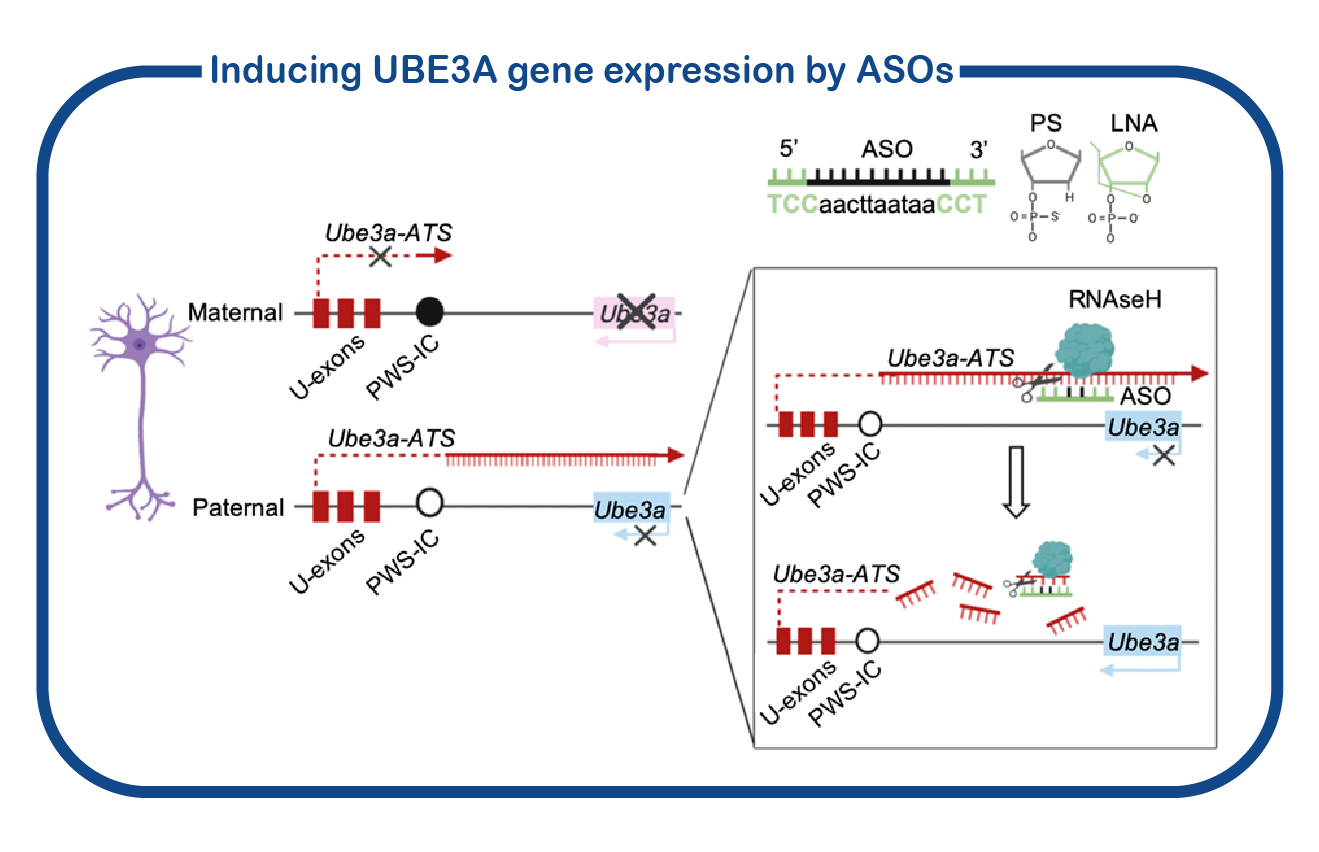
Lecoquierre F, et.al. (2024) A recurrent missense variant in the E3 ubiquitin ligase substrate recognition subunit FEM1B causes a rare syndromic neurodevelopmental disorder. Genet Med. Pubmed
Hagenaar DA, et.al. (2024) Outcome measures in Angelman syndrome. J Neurodev Disord. Pubmed
Hagenaar DA, et.al. (2023) Child characteristics associated with child quality of life and parenting stress in Angelman syndrome. J Intellect Disabil Res. Pubmed
Bindels-de Heus KGCB, et.al. (2023) Bone health in children with Angelman syndrome at the ENCORE Expertise Center. Eur J Pediatr. Pubmed
Bindels-de Heus KGCB, et.al. (2023) Hyperphagia, Growth, and Puberty in Children with Angelman Syndrome. J Clin Med. Pubmed
Keary C, et.al. (2023) Gaboxadol in angelman syndrome: A double-blind, parallel-group, randomized placebo-controlled phase 3 study. Eur J Paediatr Neurol. Aug 1;47:6-12. Pubmed
Rotaru DC, et.al. (2023). UBE3A expression during early postnatal brain development is required for proper dorsomedial striatal maturation. JCI Insight. Feb 22;8(4):e166073. Pubmed
Bindels-deHeus KGCB, et.al. (2023) Sleep problems in children with Angelman Syndrome: The effect of a behavioral intervention program. Res Dev Disabil. Feb 6;135:104444. Pubmed
Viho EMG, et.al. (2022) The Hippocampal Response to Acute Corticosterone Elevation Is Altered in a Mouse Model for Angelman Syndrome. Int J Mol Sci. Dec 24;24(1):303. Pubmed
Tanas JK, et.al. (2022) Multidimensional analysis of behavior predicts genotype with high accuracy in a mouse model of Angelman syndrome. Psychiatry. Pubmed
Lubbers K, et.al. (2022) Autism Symptoms in Children and Young Adults With Fragile X Syndrome, Angelman Syndrome, Tuberous Sclerosis Complex, and Neurofibromatosis Type 1: A Cross-Syndrome Comparison. Front Psychiatry. Pubmed
Zampeta FI, Distel B, Elgersma Y, Iping R. (2022) From first report to clinical trials: a bibliometric overview and visualization of the development of Angelman syndrome research. Hum Genet. Pubmed
Pandya NJ, et.al. (2022) A cross-species spatiotemporal proteomic analysis identifies UBE3A-dependent signaling pathways and targets. Mol Psychiatry. Pubmed
Duis J, et.al. (2022) A multidisciplinary approach and consensus statement to establish standards of care for Angelman syndrome. Mol Genet Genomic Med. e1843 Pubmed
Judson MC, el.al. (2021) Dual-isoform hUBE3A gene transfer improves behavioral and seizure outcomes in Angelman syndrome model mice. JCI Insight 6(20):e144712. Pubmed
Pandya NJ, et.al. (2021) Secreted retrovirus-like GAG-domain-containing protein PEG10 is regulated by UBE3A and is involved in Angelman syndrome pathophysiology. Cell Rep Med. 2(8):100360. Pubmed
Milazzo C, Mientjes EJ, et.al. (2021) Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. JCI Insight Aug 9;6(15):145991. Pubmed
Bossuyt S, et.al. (2021) Loss of nuclear UBE3A activity is the predominant cause of Angelman syndrome in individuals carrying UBE3A missense mutations. Hum Mol Genet. Pubmed
Elgersma Y & Sonzogni M. (2021) UBE3A reinstatement as a disease-modifying therapy for Angelman syndrome. Dev Med Child Neurol. Pubmed
Avagliano Trezza T, et.al. (2021) Mono-ubiquitination of Rhabphilin 3A by UBE3A serves a non-degradative function. Sci Rep. 11(1):3007. Pubmed
Den Besten I, et.al. (2020) Clinical aspects of a large group of adults with Angelman syndrome. Am J Med Genet A. Pubmed
Sonzogni M, et.al. (2020) Assessing the requirements of prenatal UBE3A expression for rescue of behavioral phenotypes in a mouse model for Angelman syndrome. Mol Autism. 11(1):70. Pubmed
Geerts-Haages A, et.al. (2020) A novel UBE3A sequence variant identified in eight related individuals with neurodevelopmental delay, results in a phenotype which does not match the clinical criteria of Angelman syndrome. Mol Genet Genomic Med. Pubmed
Zampeta IF, et.al. (2020) Conserved UBE3A subcellular distribution between human and mice is facilitated by non-homologous isoforms. Hum Mol Genet. Pubmed
Bindels-de Heus KGCB, et al. (2020) An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. 182:53–63. Pubmed
Rotaru DC et.al. (2020) Angelman Syndrome: From Mouse Models to Therapy. Neuroscience. 4522:30103-2 . Pubmed
Sonzogni M, et al. (2019) Delayed loss of UBE3A reduces the expression of Angelman syndrome-associated phenotypes. Mol Autism. Pubmed
Tonazzini I, et.al. (2019) The role of ubiquitin ligase E3A in polarized contact guidance and rescue strategies in UBE3A-deficient hippocampal neurons. Mol Autism. Pubmed
Avagliano Trezza R, et al. (2019) Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat Neurosci. 22; 1235–47. Pubmed
Wang T, et.al. (2018) Enhanced transmission at the calyx of held synapse in a mouse model for angelman syndrome. Front Cell Neurosci. Pubmed
Rotaru DC, et.al. (2018) Adult Ube3a gene reinstatement restores the electrophysiological deficits of prefrontal cortex layer 5 neurons in a mouse model of angelman syndrome. J Neurosci. 38; 8011–30. Pubmed
Sonzogni M, et.al. (2018) A behavioral test battery for mouse models of Angelman syndrome: A powerful tool for testing drugs and novel Ube3a mutants. Mol Autism. 14; 9-47. Pubmed
Judson MCC, et al. (2016) GABAergic Neuron-Specific Loss of Ube3a Causes Angelman Syndrome-Like EEG Abnormalities and Enhances Seizure Susceptibility. Neuron. 90; 56–69. Pubmed
Tonazzini I, et.al. (2016) Impaired Neurite Contact Guidance in Ubiquitin Ligase E3a (Ube3a)-Deficient Hippocampal Neurons on Nanostructured Substrates. Adv Healthc Mater 5; 850–62. Pubmed
Elgersma Y. (2015) Neurodevelopmental disease: A molecular tightrope. Nature 526; 50–1. Pubmed
Silva-Santos S, et.al. (2015) Ube3a reinstatement identifies distinct developmental windows in a murine Angelman Syndrome model. J Clin Invest. 125; 2069-76. Pubmed
Steinkellner, T. et al. (2012) Ca(2+)/calmodulin-dependent protein kinase IIα (αCaMKII) controls the activity of the dopamine transporter: implications for Angelman syndrome. J Biol Chem 287, 29627–29635. Pubmed
van Woerden, G.M. et al. (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci 10, 280–282. Pubmed
Elgersma, Y. (2007) Genetic engineering cures mice of neurological deficits: prospects for treating Angelman syndrome. Pharmacogenomics 8, 539–541. Pubmed
van den Ouweland, A.M. et al. (1999) Angelman syndrome: AS phenotype correlated with specific EEG pattern may result in a high detection rate of mutations in the UBE3A gene. J Med Genet 36, 723–724. Pubmed
Fang, P. et al. (1999) The spectrum of mutations in UBE3A causing Angelman syndrome. Hum Mol Genet 8, 129–135. Pubmed
Buiting, K. et al. (1998) Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counseling, and prenatal diagnosis. Am J Hum Genet 63, 170–180. Pubmed
Horsthemke, B. et al. (1996) Familial translocations involving 15q11-q13 can give rise to interstitial deletions causing Prader-Willi or Angelman syndrome. J Med Genet 33, 848–851. Pubmed
van den Ouweland, A.M. et al. (1995) DNA diagnosis of Prader-Willi and Angelman syndromes with the probe PW71 (D15S63). Hum Genet 95, 562–567. Pubmed