SYNGAP1 Syndrome is an autosomal dominant genetic disorder caused by mutations that cause a loss-of-function of the protein called SYNGAP1. Since its discovery, the number of patients diagnosed with a SYNGAP1 mutation has rapidly increased. It is now estimated that 1% of all individuals with intellectual disability carry a mutation in the SYNGAP1 gene.
SYNGAP1 belongs to a large group of disorders referred to as RASopathies. These proteins are forming a signaling cascade in the cell (depicted in red in the figure). The central nodes of this signaling pathway are formed by the RAS proteins. However, whereas most proteins of the RASopathies are present in all cells in the body, the SYNGAP1 protein is exclusively present in the brain, and more specifically, in the synapse. Therefore, the phenotype of SYNGAP1 patients is restricted to neurological problems and distinctively different from other RASopathies in which there are also many non-neuronal problems.
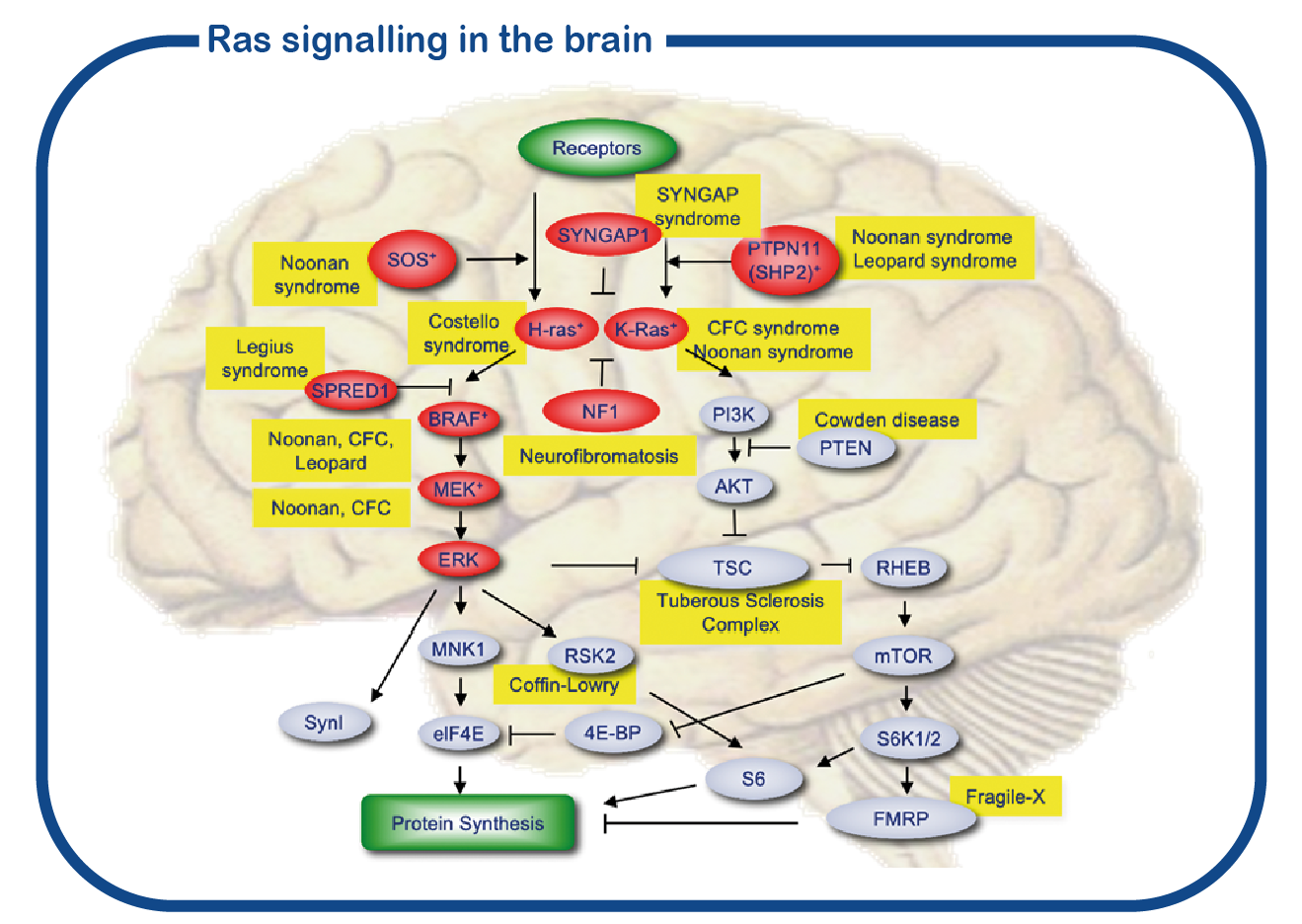
SYNGAP1 syndrome is associated with intellectual disability, an increased risk of epilepsy, difficult to understand behavior and various physical complaints, such as gastrointestinal problems, movement problems and sleep problems. These problems can seriously affect the quality of life of the patient and relatives/caregivers. Too little is currently known about the course of the condition in adulthood. In collaboration with the Adult Genetic Epilepsy Clinic of the Toronto Western Hospital in Canada, we have started a multicenter study, in which both adult patients from Canada and the Netherlands will be examined. The SYNGAP1 expertise center is a collaboration within ENCORE of the Erasmus MC and the Zuidwester Foundation. Our goal is to better understand the course of SYNGAP1 syndrome in adulthood. We will also study which genetic factors influence the risk of epilepsy and difficult to understand behavior in people with SYNGAP1 syndrome.
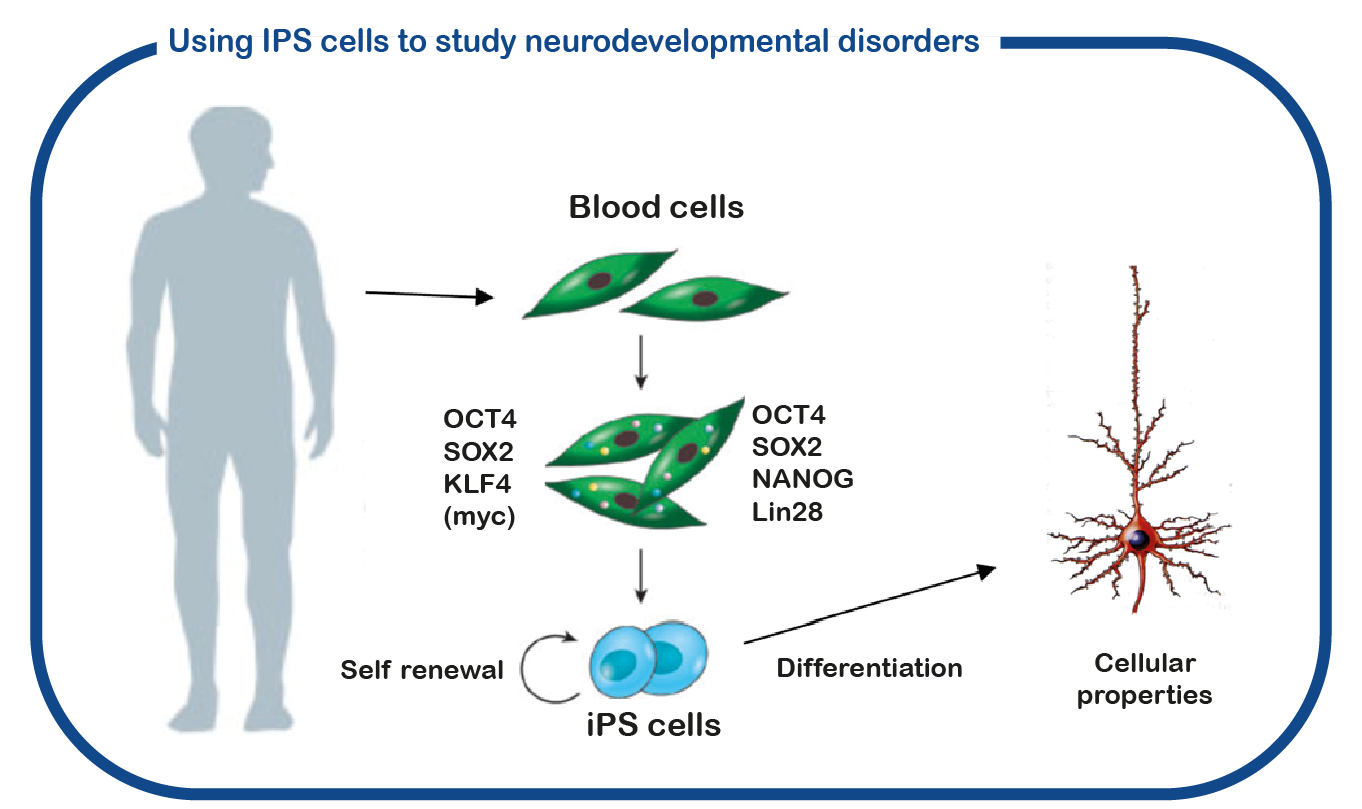
SYNGAP1 syndrome can be considered a so-called RASopathy. At ENCORE we study RASopathies mainly by studying NF1 and Costello syndrome. We mainly focus on the cognitive deficits associated with increased activity of the RAS signaling pathway. For this we use the Nf1 and Costello mouse model (Omrani, Mol Psychiatry, 2015; Schreiber, Sci Rep, 2017). We use cognitive testing to investigate the learning deficits and to try to improve these deficits with drugs that target the RAS signaling pathway. In addition to using mouse models, we also use induced pluripotent stem cells (iPS). These stem cells are generated from blood cells donated by patients and unaffected family members. The major advantage of such iPS cells is that we can differentiate these cells into human neurons, allowing us to study human (patient) neurons in a culture dish. With this approach we hope to decipher how increased RAS activity disrupts the brain and to identify new drugs that restore brain cell function.
Vlaskamp DRM, et.al. (2019) SYNGAP1 Encephalopathy: A Distinctive Generalized Developmental and Epileptic Encephalopathy. Neurology. Pubmed
Schreiber J, et al. (2017) Mechanisms underlying cognitive deficits in a mouse model for Costello Syndrome are distinct from other RASopathy mouse models. Sci Rep. Pubmed
Omrani A, et al. (2015) HCN channels are a novel therapeutic target for cognitive dysfunction in Neurofibromatosis type 1. Mol Psychiatry 20; 1311–21. Pubmed