SYNGAP1-syndroom is een autosomaal dominante genetische aandoening die wordt veroorzaakt door mutaties die een functieverlies veroorzaken van het eiwit SYNGAP1. Sinds de ontdekking ervan is het aantal patiënten met de diagnose SYNGAP1-mutatie snel toegenomen. Er wordt nu geschat dat 1% van alle personen met een verstandelijke beperking een mutatie in het SYNGAP1-gen draagt.
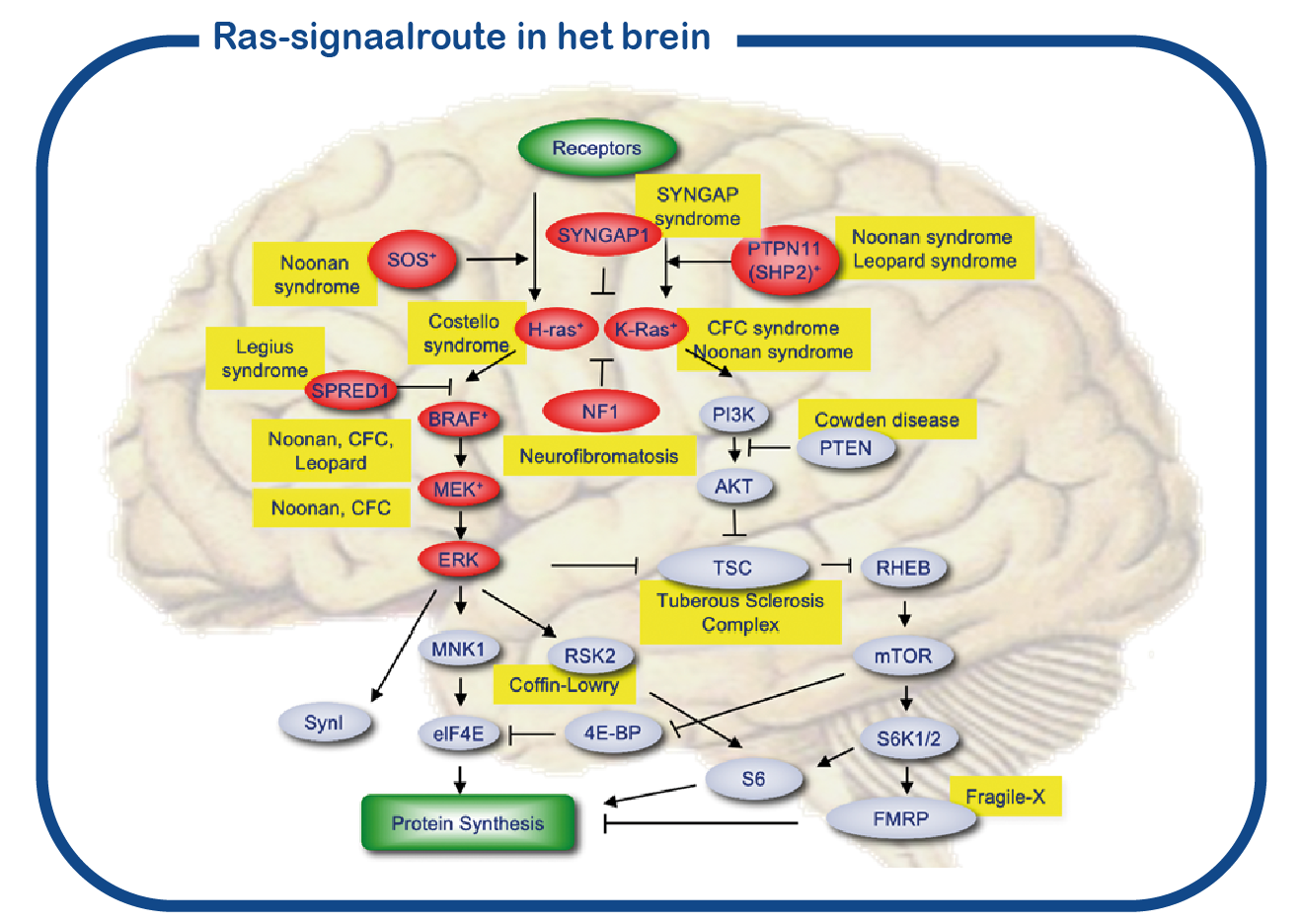
SYNGAP1 behoort tot een grote groep aandoeningen die RASopathieën worden genoemd. Deze eiwitten vormen een signaalcascade in de cel (weergegeven in rood in de figuur). De centrale knooppunten van deze signaalroute worden gevormd door de RAS-eiwitten. Hoewel de meeste eiwitten van de RASopathieën in alle cellen in het lichaam aanwezig zijn, is het SYNGAP1-eiwit uitsluitend aanwezig in de hersenen, en meer specifiek in de synaps. Daarom is het fenotype van SYNGAP1-patiënten beperkt tot neurologische problemen en onderscheidend van andere RASopathieën waarbij er ook veel niet-neuronale problemen zijn.
SYNGAP1-syndroom is geassocieerd met een verstandelijke beperking, een verhoogd risico op epilepsie, moeilijk verstaanbaar gedrag en verschillende lichamelijke klachten, zoals maag-darmproblemen, bewegingsproblemen en slaapproblemen. Deze problemen kunnen de kwaliteit van leven van de patiënt en van de verwanten/ verzorgers sterk beïnvloeden. Er is momenteel te weinig bekend over het beloop van de aandoening op volwassen leeftijd. In samenwerking met Adult Genetic Epilepsy Clinic van het Toronto Western Hospital uit Canada hebben wij een multicenter studie opgestart, waarbij zowel volwassen patiënten uit Canada als Nederland zullen worden onderzocht. Het SYNGAP1 expertisecentrum is een samenwerking binnen ENCORE van het Erasmus MC en Stichting Zuidwester. Ons doel is om beter te begrijpen wat het beloop van het SYNGAP1-syndroom op volwassen leeftijd is. Ook zullen we bestuderen welke genetische factoren het risico op epilepsie en moeilijk verstaanbaar gedrag beïnvloeden bij mensen met SYNGAP1-syndroom.
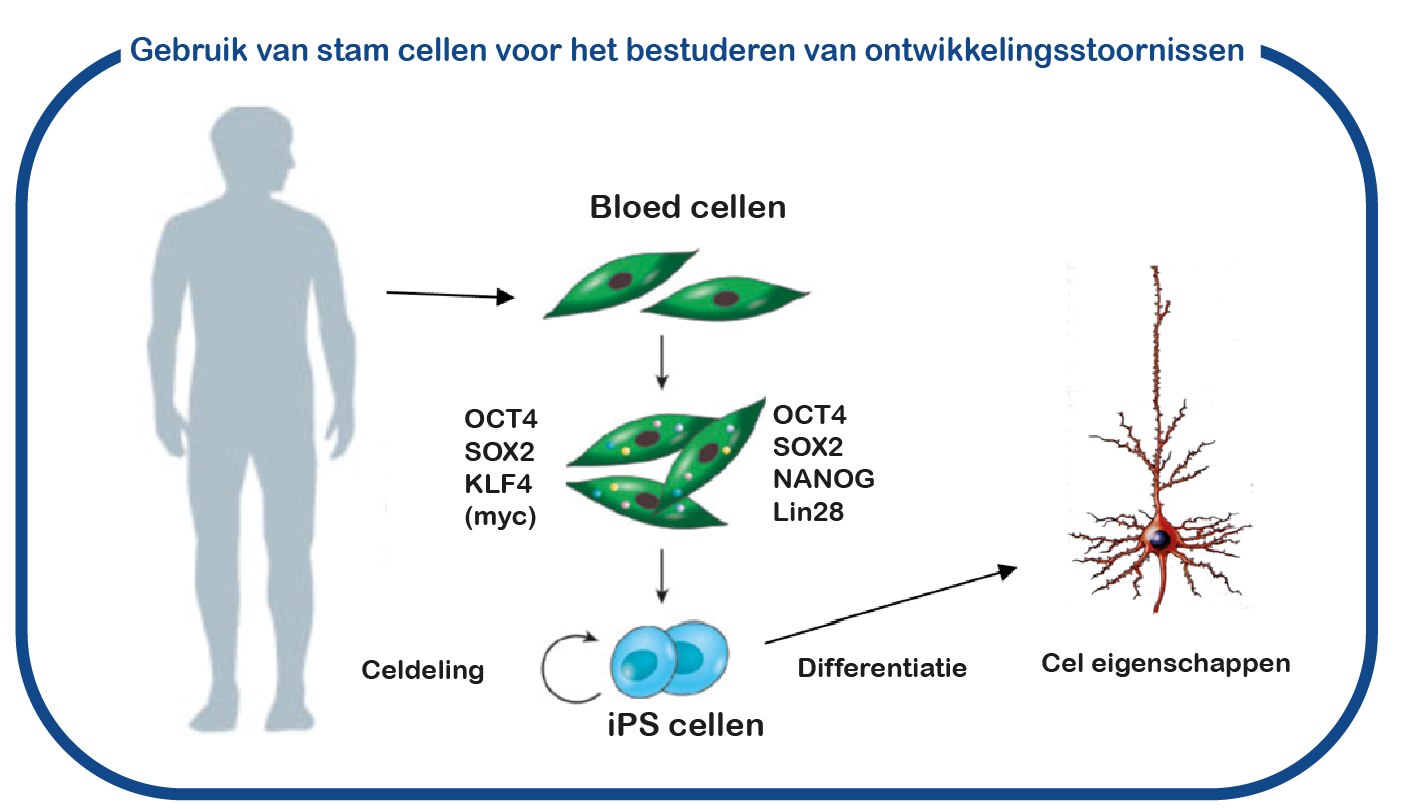
SYNGAP1-syndroom kan worden beschouwd als een zogenaamde RASopathie. Bij ENCORE bestuderen we de RASopathieën voornamelijk door NF1 en Costello syndroom te bestuderen. We richten ons voornamelijk op de cognitieve gebreken die geassocieerd zijn met verhoogde activiteit van de RAS-signaalroute. Daarvoor maken we gebruik van het Nf1 en Costello muismodel (Omrani, Mol Psychiatry, 2015; Schreiber, Sci Rep, 2017). We gebruiken cognitieve testen om de leerproblemen te onderzoeken en om te proberen deze tekorten te verbeteren met geneesmiddelen die op het RAS- signaalroute zijn gericht. Naast het gebruik van muismodellen gebruiken we ook geïnduceerde pluripotente stamcellen (iPS). Deze stamcellen worden gegenereerd uit bloedcellen die zijn gedoneerd door patiënten en niet-aangedane familieleden. Het grote voordeel van dergelijke iPS-cellen is dat we deze cellen kunnen differentiëren tot menselijke neuronen, waardoor we menselijke (patiënt) neuronen in een kweekschaal kunnen bestuderen. Met deze benadering hopen we te ontcijferen hoe verhoogde RAS activiteit de hersenen ontregelt en om nieuwe geneesmiddelen te identificeren die de hersencel functie herstelt.
Vlaskamp DRM, et.al. (2019) SYNGAP1 Encephalopathy: A Distinctive Generalized Developmental and Epileptic Encephalopathy. Neurology. Pubmed
Schreiber J, et al. (2017) Mechanisms underlying cognitive deficits in a mouse model for Costello Syndrome are distinct from other RASopathy mouse models. Sci Rep. Pubmed
Omrani A, et al. (2015) HCN channels are a novel therapeutic target for cognitive dysfunction in Neurofibromatosis type 1. Mol Psychiatry 20; 1311–21. Pubmed