Tubereuze sclerosecomplex (TSC) is een genetische aandoening die veel verschillende organen aantast, waaronder de ogen, het hart, de nieren, de huid, de longen en de hersenen. Hoewel de ernst van de symptomen van individuele patiënten sterk varieert, hebben TSC-symptomen die verband houden met de hersenen meestal het sterkste effect op de kwaliteit van leven. De meeste patiënten krijgen epileptische aanvallen, ontwikkelingsachterstand, verstandelijke beperking en autisme. Schattingen geven aan dat een op de 6000 geboren kinderen wordt getroffen door TSC. Veel gevallen blijven echter niet gediagnosticeerd als gevolg van milde symptomen en sommige TSC-patiënten blijven onopgemerkt totdat een kind of broer of zus de diagnose TSC krijgt. Een derde van de patiënten erft de genetische mutatie van een (licht aangetaste) ouder, terwijl twee derde de mutatie ‘de novo’ ontwikkelt, wat betekent dat de mutatie spontaan ontstaat bij de foetus.
TSC wordt veroorzaakt door een mutatie in het TSC1- of het TSC2-gen, dat codeert voor respectievelijk de eiwitten hamartin en tuberin. Deze eiwitten werken samen om de activiteit van het mTOR-enzym te reguleren. Een mutatie in een van beide genen zorgt ervoor dat mTOR hyperactief wordt, wat verschillende cruciale functies in de neuron beïnvloedt.
In het lab zijn we vooral geïnteresseerd in het begrijpen van de hersen=gerelateerde symptomen van TSC. Daarbij zijn twee hoofdlijnen te onderscheiden; het ene richt zich op de mechanismen van TSC-gerelateerde epilepsie, en het andere op het begrijpen waarom de ernst van de ziekte zo divers is.
Epilepsie onderzoek.
Om het epileptische brein te bestuderen, gebruiken we een muismodel van TSC. Ons onderzoek richt zich met name op het begrijpen van het proces dat de ontwikkeling van epilepsie veroorzaakt, een proces dat epileptogenese wordt genoemd. Het analyseren van de neuronen zal ons helpen dit proces te begrijpen. Het voordeel van ons modelsysteem schuilt in een genetische truc: TSC1 gen kan op elk gewenst moment worden uitgeschakeld. Hierdoor kunnen we nauwkeurig de cellulaire en moleculaire veranderingen bestuderen die worden veroorzaakt door het verlies van het TSC-gen en uiteindelijk epilepsie veroorzaken. Door dit proces te begrijpen, hopen we betere medicijnen te ontwikkelen. We gebruiken deze muizen ook om te testen welke anti-epileptica het beste werken voor de behandeling van TSC-gerelateerde epilepsie (Koene, Annals of Clinical and Translational Neurology, 2019).
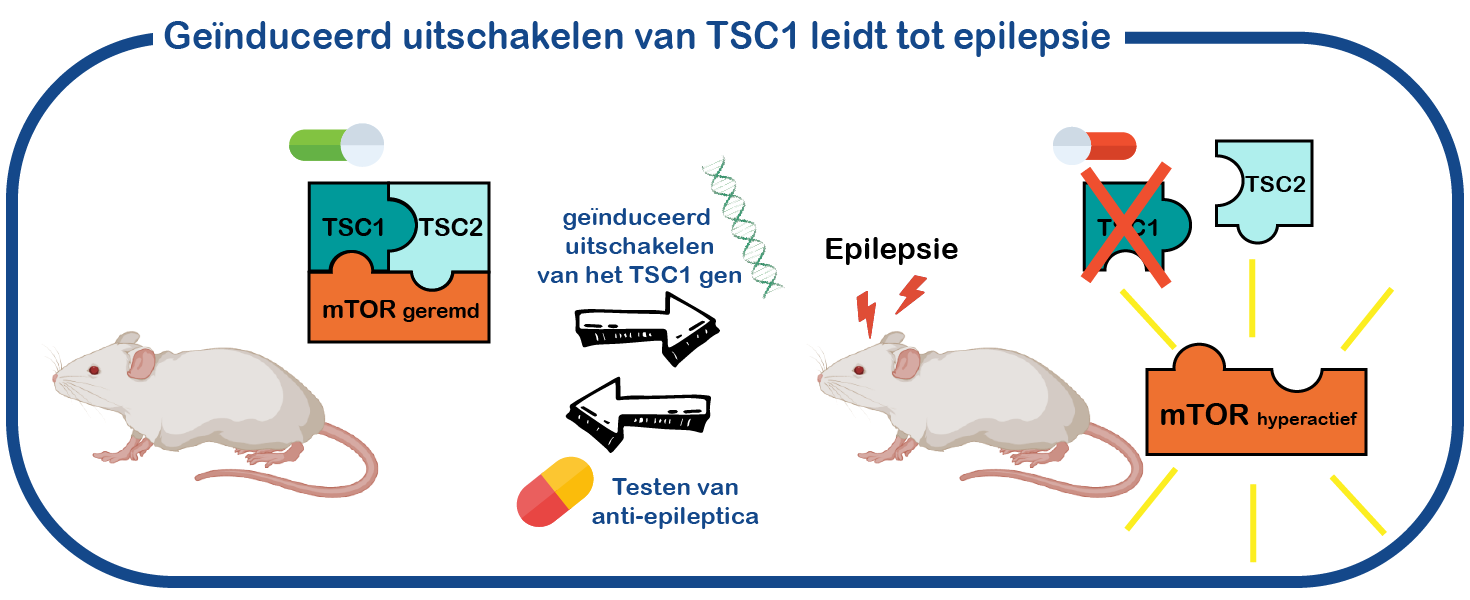
Variatie in ernst van TSC.
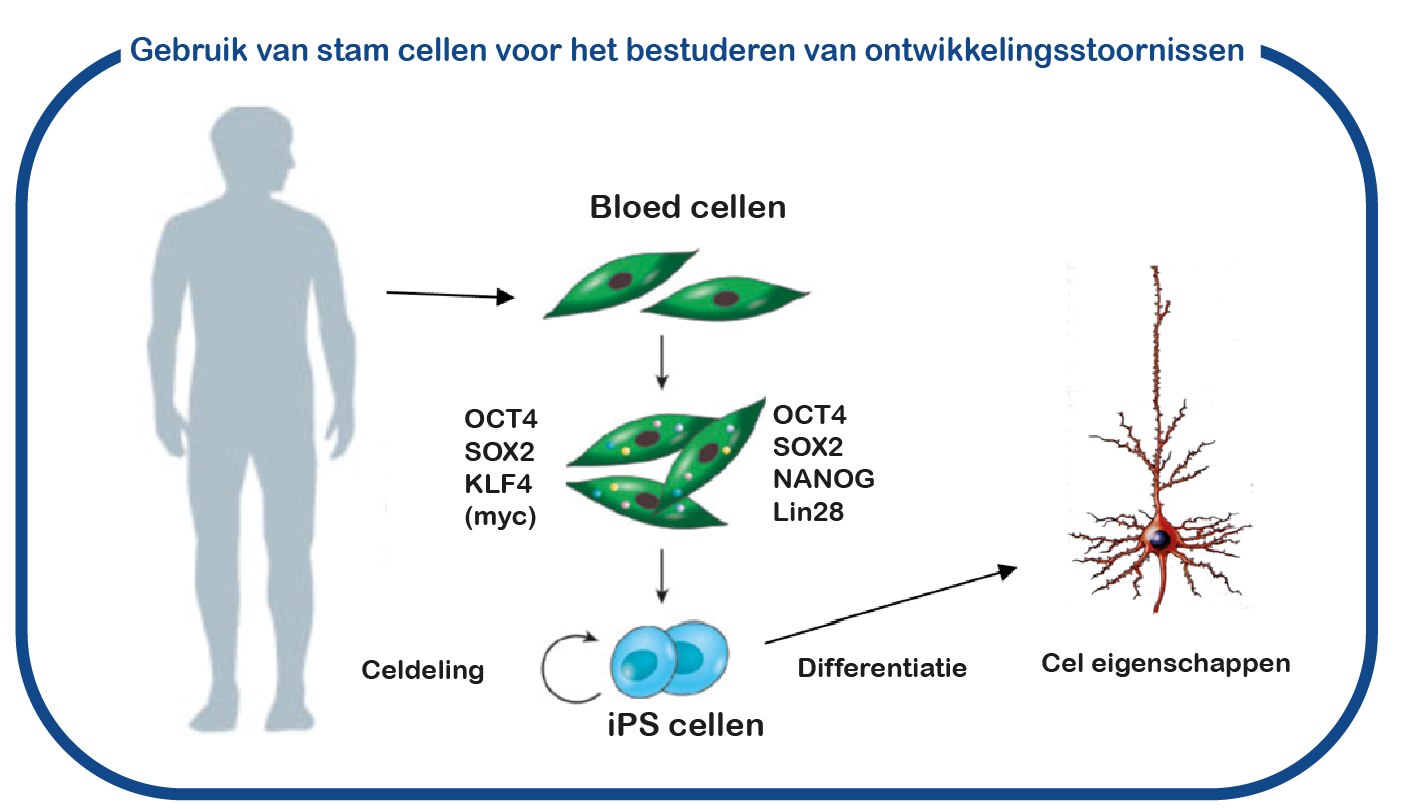
Over het algemeen vertonen TSC-patiënten met een mutatie in het TSC2-gen ernstigere symptomen dan degenen zonder TSC1. Maar zelfs als dezelfde mutatie van ouder op kind wordt overgeërfd, kunnen hun symptomen nog steeds sterk uiteenlopen. Als we de onderliggende oorzaken van deze verschillen begrijpen, kunnen we nieuwe behandelingen ontwikkelen. Om dit te bestuderen kunnen we geen muizen gebruiken, omdat deze allemaal genetisch identiek zijn en niet zo’n variabiliteit in symptomen vertonen. Daarom gebruiken we geïnduceerde pluripotente stamcellen (iPS). Deze stamcellen worden gegenereerd uit bloedcellen die zijn gedoneerd door patiënten en niet-aangedane familieleden. Het grote voordeel van dergelijke iPS-cellen is dat we deze cellen kunnen differentiëren tot menselijke neuronen, waardoor we menselijke (patiënt) neuronen in een kweekschaal kunnen bestuderen. Met deze aanpak hopen we factoren te ontcijferen die de variatie in symptomen veroorzaken. Deze zouden de ernst van de te ontwikkelen TSC-symptomen in een vroeg stadium kunnen voorspellen, en mogelijk de sleutel geven tot vroege behandelingen, zodat we de kwaliteit van leven kunnen verbeteren.
Heuvelmans AM, et.al. (2024) Modeling mTORopathy-related epilepsy in cultured murine hippocampal neurons using the multi-electrode array. Exp Neurol. Pubmed
Müller AR, et.al. (2024) Cannabidiol (Epidyolex®) for severe behavioral manifestations in patients with tuberous sclerosis complex, mucopolysaccharidosis type III and fragile X syndrome: protocol for a series of randomized, placebo-controlled N-of-1 trials. BMC Psychiatry. Pubmed
Müller AR, et.al. (2023) Understanding the impact of tuberous sclerosis complex: development and validation of the TSC-PROM. BMC Med. Aug 8;21(1):298. Pubmed
Lubbers K, et.al. (2022) Autism Symptoms in Children and Young Adults With Fragile X Syndrome, Angelman Syndrome, Tuberous Sclerosis Complex, and Neurofibromatosis Type 1: A Cross-Syndrome Comparison. Front Psychiatry. Pubmed
Koene LM, et.al. (2021) Identifying the temporal electrophysiological and molecular changes that contribute to TSC-associated epileptogenesis. JCI Insight. 6(23):e150120. Pubmed
Koene LMC, et al. (2019) Effects of antiepileptic drugs in a new TSC/mTOR-dependent epilepsy mouse model. Ann Clin Transl Neurol. 6; 1273–91. Pubmed
Overwater IE, et al. (2019) A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology. 93; E200–9. Pubmed
Overwater IE, et.al. (2019) Everolimus for the treatment of refractory seizures associated with tuberous sclerosis complex (TSC): Current perspectives. Ther Clin Risk Manag. 951–5. Pubmed
Mous SE, et al. (2018) Cortical dysplasia and autistic trait severity in children with Tuberous Sclerosis Complex: a clinical epidemiological study. Eur Child Adolesc Psychiatry. 27; 753–65. Pubmed
Both P, et al. (2018) Tuberous sclerosis complex: Concerns and needs of patients and parents from the transitional period to adulthood. Epilepsy Behav. Pubmed
Overwater IE, et al. (2017) Interdependence of clinical factors predicting cognition in children with tuberous sclerosis complex. J Neurol. 264; 161–7. Pubmed
Reijnders MRF, et al. (2017) Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat Commun. Pubmed
Overwater IE, et al. (2016) Genotype and brain pathology phenotype in children with tuberous sclerosis complex. Eur J Hum Genet. 24; 1688–95. Pubmed
Overwater IE, et al. (2016) Sirolimus for epilepsy in children with tuberous sclerosis complex. Neurology. 87; 1011–8. Pubmed
Overwater IE, et al. (2015) Epilepsy in children with tuberous sclerosis complex: Chance of remission and response to antiepileptic drugs. Epilepsia. 56; 1239–45. Pubmed
Goorden SMI, et.al. (2015) Intact neuronal function in Rheb1 mutant mice: implications for TORC1-based treatments. Hum Mol Genet 24; 3390–8. Pubmed
Peters JM, et al. (2014) Diffusion tensor imaging and related techniques in tuberous sclerosis complex: review and future directions. Future Neurol 8; 583–97. Pubmed
Overwater IE, et.al. (2014) Treatment of intractable epilepsy in tuberous sclerosis complex with everolimus is not yet evidence-based. Ann Neurol. Pubmed
Abs, E. et al. (2013) TORC1-dependent epilepsy caused by acute biallelic Tsc1 deletion in adult mice. Ann Neurol 74, 569–579. Pubmed
Overwater, I.E. et al. (2013) Behandelingen voor genetische neurocognitieve aandoeningen. Neuropraxis 5, 132–138. Link
van Eeghen, A.M. et al. (2013) The neuroanatomical phenotype of tuberous sclerosis complex: focus on radial migration lines. Neuroradiology 55, 1007–1014. Pubmed
Melser, S. et al. (2013) Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metabolism 17, 719–730. Pubmed
van Eeghen, A.M. et al. (2013) Central TSC2 missense mutations are associated with a reduced risk of infantile spasms. Epilepsy Res 103, 83–87. Pubmed
van Eeghen, A.M. et al. (2012) Understanding relationships between autism, intelligence, and epilepsy: a cross-disorder approach. Dev Med Child Neurol 55, 146–153. Pubmed
Hoogeveen-Westerveld, M. et al. (2012) Functional Assessment of TSC2 Variants Identified in Individuals with Tuberous Sclerosis Complex. Hum Mutat. Pubmed
van Eeghen, A.M. et al. (2012) Genotype and cognitive phenotype of patients with tuberous sclerosis complex. Eur J Hum Genet 20, 510–515. Pubmed
van Eeghen, A.M. et al. (2012) Cognitive and adaptive development of patients with tuberous sclerosis complex: A retrospective, longitudinal investigation. Epilepsy Behav 23, 10–15. Pubmed
Goorden, S.M.I. et al. (2011) Rheb is essential for murine development. Mol Cell Biol 31, 1672–1678. Pubmed
Goorden, S.M.I. and Elgersma, Y. (2011) Rheb: enrichment beyond the brain. Cell Cycle 10, 2412–2413. Pubmed
van den Ouweland, A.M.W. et al. (2011) Characterisation of TSC1 promoter deletions in tuberous sclerosis complex patients. Eur J Hum Genet 19, 157–163. Pubmed
van Eeghen, A.M. et al. (2011) Characterizing sleep disorders of adults with tuberous sclerosis complex: a questionnaire-based study and review. Epilepsy Behav 20, 68–74. Pubmed
Goorden, S.M.I. et al. (2007) Cognitive deficits in Tsc1+/- mice in the absence of cerebral lesions and seizures. Ann Neurol 62, 648–655. Pubmed
Sancak, O. et al. (2005) Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet 13, 731–741. Pubmed
Nellist, M. et al. (2005) Large deletion at the TSC1 locus in a family with tuberous sclerosis complex. Genet. Test. 9, 226–230. Pubmed
Nellist, M. et al. (2003) Regulation of tuberous sclerosis complex (TSC) function by 14-3-3 proteins. Biochem Soc Trans 31, 587–591. Pubmed
Nellist, M. et al. (2001) TSC2 missense mutations inhibit tuberin phosphorylation and prevent formation of the tuberin-hamartin complex. Hum Mol Genet 10, 2889–2898. Pubmed
Goedbloed, M.A. et al. (2001) Analysis of TSC2 stop codon variants found in tuberous sclerosis patients. Eur J Hum Genet 9, 823–828. Pubmed
Nellist, M. et al. (1999) Characterization of the cytosolic tuberin-hamartin complex. Tuberin is a cytosolic chaperone for hamartin. J Biol Chem 274, 35647–35652. Pubmed
Verhoef, S. et al. (1999) High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet 64, 1632–1637. Pubmed
van Slegtenhorst, M. et al. (1999) Mutational spectrum of the TSC1 gene in a cohort of 225 tuberous sclerosis complex patients: no evidence for genotype-phenotype correlation. J Med Genet 36, 285–289. Pubmed
Verhoef, S. et al. (1999) Malignant pancreatic tumour within the spectrum of tuberous sclerosis complex in childhood. Eur J Pediatr 158, 284–287. Pubmed
van Slegtenhorst, M. et al. (1998) Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet 7, 1053–1057. Pubmed
Wang, Q. et al. (1998) Identification of a large insertion and two novel point mutations (3671del8 and S1221X) in tuberous sclerosis complex (TSC) patients. Mutations in brief no. 119. Online. Hum Mutat 11, 331–332. Pubmed
van Slegtenhorst, M. et al. (1997) Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808. Pubmed
Rinke de Wit, T.F. et al. (1996) Expression of tyrosine kinase gene in mouse thymic stromal cells. Int Immunol. 8, 1787–1795. Pubmed
Vrtel, R. et al. (1996) Identification of a nonsense mutation at the 5′ end of the TSC2 gene in a family with a presumptive diagnosis of tuberous sclerosis complex. J Med Genet 33, 47–51. Pubmed
Halley, D.J. (1996) Tuberous sclerosis: between genetic and physical analysis. Acta Genet Med Gemellol (Roma) 45, 63–75. Pubmed
van Slegtenhorst, M. et al. (1995) Cosmid contigs from the tuberous sclerosis candidate region on chromosome 9q34. Eur J Hum Genet 3, 78–86. Pubmed
Janssen, B. et al. (1994) Refined localization of TSC1 by combined analysis of 9q34 and 16p13 data in 14 tuberous sclerosis families. Hum Genet 94, 437–440. Pubmed