Om de best mogelijke zorg te verlenen aan patiënten met een zeldzame aandoening is het essentieel dat er wereldwijd kennis over de aandoening vergaard wordt. Om de zorg voor zeldzame aandoeningen en het vergaren van kennis te stimuleren zijn er landelijke expertisecentra opgezet. Voor de formele erkenning van een expertisecentrum door het ministerie van VWS, is een belangrijke voorwaarde dat het expertisecentrum kennis vergaart, analyseert en deelt door middel van publicaties. Dit kunnen publicaties zijn in wetenschappelijke tijdschriften, maar ook behandel richtlijnen voor artsen of informatie brochures voor patiënten of verzorgers. Door gestandaardiseerde follow-up, en nauwe samenwerking tussen artsen en onderzoekers optimaliseren we de zorg en het onderzoek binnen ENCORE. Op die manier kunnen we uiteindelijk betere behandelingen ontwikkelen voor zeldzame aandoeningen. U kunt daarom gevraagd worden om deel te nemen aan onderzoek. Deelname aan onderzoek is altijd op vrijwillige basis. De verkregen data wordt in anonieme vorm opgeslagen en geanalyseerd. Al het onderzoek is vooraf goedgekeurd door een ethische toetsingscommissie.
Bij alle Angelman Syndroom patiënten die in ons expertisecentrum gezien worden zal genetisch onderzoek worden verricht om de genetische oorzaak vast te stellen en de ouders te kunnen ondersteunen en adviseren. Indien dit genetisch onderzoek elders al gebeurd is, zal dit niet opnieuw gedaan worden. Deze genetische kennis helpt ons ook om het effect van de genetische verandering (‘mutatie’) op de ernst van de symptomen beter te begrijpen. Tevens kunnen we dan onderzoeken welke behandeling het beste werkt voor een bepaalde genetische verandering. In zeldzame gevallen is de genetische verandering niet eenduidig. In die gevallen zal de genetische verandering verder onderzocht worden op het laboratorium (zie pre-klinisch onderzoek).
Het Angelman expertisecentrum is het grootste Angelman Syndroom expertisecentrum ter wereld. Onze klinische bevindingen van 100 kinderen en 100 volwassenen met het Angelman Syndroom hebben we onlangs gepubliceerd (De Heus, 2019; Besten, 2020) .
Gedetailleerde kennis over het beloop van Angelman Syndroom (welke symptomen en klachten zijn er, en wanneer ontstaan die precies) is van groot belang om in de toekomst klachten voortijdig te kunnen herkennen en optimaal te behandelen. Ook willen we bepalen op welke manier de effectiviteit van behandelingen het beste kan worden gemeten (uitkomstmaten onderzoek). Dit is van groot belang voor geneesmiddelen onderzoek (trials). Immers, alleen als we kunnen aantonen dat een nieuw medicijn de kwaliteit van leven verbetert ten opzichte van een onbehandelde patiënt, zal het medicijn daadwerkelijk goedgekeurd en vergoed worden.
Gedragsmatige behandeling voor slaapproblemen
Veel kinderen met het Angelman syndroom hebben problemen met inslapen, doorslapen en vroeg wakker worden. In onze studie hebben we gekeken of een gedragsmatige behandeling effect heeft op de duur van slaap en het slaapgedrag. Interventies richtten zich op een duidelijk slaapritueel, een vast slaapritme en het minimaliseren van ouderlijke bezoeken aan het kind ’s nachts. Daarnaast hebben we een controlegroep 6 maanden gevolgd zonder hen een behandeling te geven. De resultaten worden op dit moment geanalyseerd en worden verwacht in het begin van volgend jaar. Ervaringen en uitkomsten van de studie worden op dit moment al doorgevoerd op de polikliniek.
Ondersteunde communicatie
Samen met de oudervereniging hebben we ons de afgelopen jaren hard gemaakt voor het stimuleren van ondersteunde communicatie (OC) bij kinderen en volwassenen met Angelman Syndroom. Informatie vanuit de literatuur en ervaringen vanuit de praktijk laten zien dat kinderen en volwassenen met AS kunnen leren communiceren middels gebaren, pictogrammen en spraakcomputers, maar dat het cruciaal is dat de omgeving hier ook mee communiceert. In onze studie coachen we ouders en begeleiders van mensen met het Angelman Syndroom, om zo een taalrijke omgeving te creëren waardoor kinderen en volwassenen met AS deze taal kunnen gaan imiteren, net zoals gewone kinderen leren spreken doordat er tegen hen gepraat wordt. Door de coronamaatregelen heeft de studie vertraging opgelopen, maar de trajecten worden naar verwachting eind 2020 afgerond. Meer informatie over OC is te vinden op www.isaac-nf.nl. Om de Leidraad Ondersteunde Communicatie waaraan ENCORE heeft bijgedragen te downloaden, klik hier.
De validiteit en betrouwbaarheid van cognitieve testen (uitkomstmaten)
Voor het verbeteren van behandelingen voor Angelman Syndroom is het essentieel dat we objectief kunnen vaststellen of de behandeling effectief is. Alleen dan kan het geneesmiddel of de behandeling geregistreerd en/of vergoed worden. Zulke objectieve testen noemen we uitkomstmaten. Samen met de ‘Angelman Syndrome Clinic’ van Boston en North-Carolina (VS) hebben we de validiteit en betrouwbaarheid van de Bayley-III test onderzocht om te gebruiken voor klinische trials. De Bayley-III is een veel gebruikte ontwikkelingstest bij kinderen met een verstandelijke beperking zoals Angelman Syndroom. In deze studie hebben we data geanalyseerd van ruim 100 kinderen, waarbij bij ongeveer de helft van de kinderen de test meerdere keren is afgenomen. We hebben onderzocht of er bepaalde items voor kinderen met AS relatief makkelijk of moeilijk zijn en of de test over meerdere metingen een betrouwbaar en stabiel beeld geeft. Dit onderzoek is relevant voor de klinische praktijk en voor de aankomende klinische trials, aangezien de Bayley-III een belangrijke uitkomstmaat is. De analyses zijn inmiddels afgerond en worden spoedig gepubliceerd.
3dMD scan
Om meer inzicht te krijgen in het effect van de verschillende Angelman Syndroom mutaties op hersenvolume (grootte van de schedel) en gezichtskenmerken, maken we gebruik van een 3dMD scanner. Dit is een gezichtsscanner die gebruik maakt van twee paar camera’s op een groot statief. De camera’s maken een 180 graden 3D beeld van het gezicht met echt uitziende huidstructuur en huidskleur. Tijdens de registratie projecteren twee projectoren willekeurige lichtpatronen op het gezicht. Het apparaat gebruikt ze als houvast voor het maken van de afbeelding. Het proces is doorgaans erg kort, met een maximum van 20 seconden per registratie. Dit betekent dat het maken van een 3D foto bij elkaar opgeteld slechts een paar minuten duurt. Na het scannen genereert het systeem automatisch een 3D beeld.
ROSA studie (Rotterdam Outcome Study for children with Angelman syndrome)
Voor kinderen met het Angelman Syndroom is er een gebrek aan geschikte meetinstrumenten (uitkomstmaten). Een meetinstrument is een vragenlijst, test, of apparaat. Wat het meet, verschilt per instrument. Bij kinderen met het Angelman syndroom zijn standaard meetinstrumenten vaak minder geschikt. Problemen met het praten, bewegen, of de concentratie verstoren de resultaten op zo’n test. Helaas is er nog maar weinig bekend over wat dan wél geschikte en haalbare meetinstrumenten zijn voor kinderen met het Angelman syndroom.
Daarom is het doel van dit onderzoek om geschikte en haalbare meetinstrumenten te vinden voor kinderen met het Angelman syndroom. Deze kunnen worden gebruikt voor (medicijn)onderzoek, en in de toekomst misschien ook in de zorg. Een tweede doel van dit onderzoek is om informatie te verzamelen over het natuurlijke beloop van het Angelman syndroom. Dat zal bijdragen aan onze kennis van deze zeldzame en complexe ziekte. Onderwerpen waar we ons in deze studie op focussen zijn: hersenactiviteit, sociale functies, voeding en stofwisseling, slaap, beweging en lopen, en hormoonhuishouding.
De ROSA studie is inmiddels afgerond. De resultaten kun je lezen in onze publicatie, klik hier deze te openen.

Klinische trials voor nieuwe behandelingen voor Angelman Syndroom
Gaboxadol
Met het ENCORE expertise centrum hebben we deelgenomen aan de NEPTUNE studie van OVID. In een eerdere klinische studie (de STARS trial) werd een positief effect gevonden van gaboxadol op het dagelijks functioneren van kinderen met Angelman Syndroom (klik hier voor meer informatie). In het ENCORE onderzoekslaboratorium werd een positief effect gevonden op de motorische vaardigheden in een muis model van het Angelman Syndroom. De NEPTUNE studie is een gerandomiseerde, placebo gecontroleerde studie waaraan 4 Nederlandse kinderen deel hebben deelgenomen. Helaas waren de verbeteringen die werden gezien gelijk voor de groep kinderen die gaboxadol toegediend kregen als de groep kinderen die het placebo ontvingen. Klik hier voor meer informatie over de resultaten. De studie is recentelijk gestopt. De resultaten laten zien hoe belangrijk het is om een interventiegroep te vergelijken met een groep patiënten die en placebo ontvangt. Op deze manier kan namelijk betrouwbaar bewijs worden geleverd over de klinische effectiviteit van een mogelijk nieuw medicijn. We willen alle deelnemende families heel erg bedanken voor hun bijdrage aan de studie.
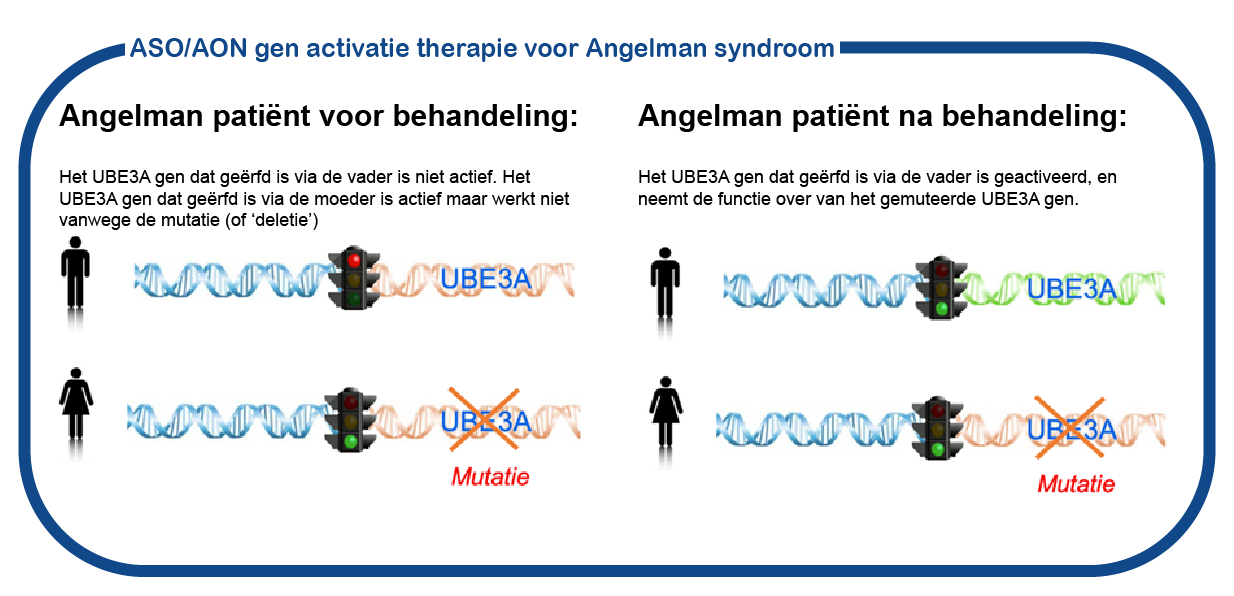
Tangelo studie
Op dit moment neemt het ENCORE expertise centrum deel aan de TANGELO studie van Roche. In deze grote, internationale fase 1/2 klinische studie ontvangen kinderen tussen de leeftijd 1 en 12 jaar oud een behandeling met een antisense oligonucleotide, een mogelijk medicijn dat is ontworpen om de kopie van het UBE3A-gen van de vader aan te zetten. Klik hier voor meer informatie over de studie. Heeft u vragen over deze studie of wilt u meer informatie, stuur ons een e-mail via angelman@erasmusmc.nl
We houden ouders met frequente nieuwsbrieven en via de website van de oudervereniging op de hoogte van de nieuwste ontwikkelingen. We werken hierbij nauw samen met de vASN patiëntorganisatie. Verder is er een internationaal AS centrum samenwerkingsverband opgericht om de klinische zorg te optimaliseren en samen te werken in wetenschappelijk onderzoek: https://www.angelman.org/angelman-syndrome-clinics/
Halpin S, et al. (2025). Advancing observer-reported outcome measurement: development of the MOOD-AS for observing distress in Angelman syndrome. JPRO, Pubmed
Ten Hooven-Radstaake M, et al. (2025). Criterion Validity, Scalability and Stability of Scoring on the Bayley-III in Children With Angelman Syndrome. JIDR, Pubmed
Loix M, et al. (2025). UBE3A promotes foam cell formation and counters remyelination by targeting ABCA1 for proteasomal degradation. Commun. , Pubmed
Milazzo C, et al. (2025). UBE3A reinstatement restores behaviorand proteome in an Angelman syndrome mouse model of imprinting defects. Autism, Pubmed
Hipp JF, et al. (2025). The UBE3A-ATS antisense oligonucleotide rugonersen in children with Angelman syndrome: a phase 1 trial.Nat med , Pubmed
Navis C, et al. (2025). Language comprehension assessment using the computer-based instrument for low motor language testing (C-BiLLT) in children with Angelman syndrome. Augmentative and alternative communication, Pubmed
van Esbroeck ACM, et al. (2025). Localization of human UBE3A isoform 3 is highly sensitive to amino acid substitutions at p.Met21 position. Mol. Genet., Pubmed
Hagenaar DA, et al. (2025). Age-Related Trajectories of Autistic Traits in Children With Angelman Syndrome. Autism res., Pubmed
Lubbers K, et al. (2024). Autism Spectrum Disorder Symptom Profiles in Fragile X Syndrome, Angelman Syndrome, Tuberous Sclerosis Complex and Neurofibromatosis Type 1. Autism Dev. Disord., Pubmed
Lecoquierre F, et.al. (2024) A recurrent missense variant in the E3 ubiquitin ligase substrate recognition subunit FEM1B causes a rare syndromic neurodevelopmental disorder. Genet Med. Pubmed
Hagenaar DA, et.al. (2024) Outcome measures in Angelman syndrome. J Neurodev Disord. Pubmed
Hagenaar DA, et.al. (2023) Child characteristics associated with child quality of life and parenting stress in Angelman syndrome. J Intellect Disabil Res. Pubmed
Bindels-de Heus KGCB, et.al. (2023) Bone health in children with Angelman syndrome at the ENCORE Expertise Center. Eur J Pediatr. Pubmed
Bindels-de Heus KGCB, et.al. (2023) Hyperphagia, Growth, and Puberty in Children with Angelman Syndrome. J Clin Med. Pubmed
Keary C, et.al. (2023) Gaboxadol in angelman syndrome: A double-blind, parallel-group, randomized placebo-controlled phase 3 study. Eur J Paediatr Neurol. Aug 1;47:6-12. Pubmed
Rotaru DC, et.al. (2023). UBE3A expression during early postnatal brain development is required for proper dorsomedial striatal maturation. JCI Insight. Feb 22;8(4):e166073. Pubmed
Bindels-deHeus KGCB, et.al. (2023) Sleep problems in children with Angelman Syndrome: The effect of a behavioral intervention program. Res Dev Disabil. Feb 6;135:104444. Pubmed
Viho EMG, et.al. (2022) The Hippocampal Response to Acute Corticosterone Elevation Is Altered in a Mouse Model for Angelman Syndrome. Int J Mol Sci. Dec 24;24(1):303. Pubmed
Tanas JK, et.al. (2022) Multidimensional analysis of behavior predicts genotype with high accuracy in a mouse model of Angelman syndrome. Psychiatry. Pubmed
Lubbers K, et.al. (2022) Autism Symptoms in Children and Young Adults With Fragile X Syndrome, Angelman Syndrome, Tuberous Sclerosis Complex, and Neurofibromatosis Type 1: A Cross-Syndrome Comparison. Front Psychiatry. Pubmed
Zampeta FI, Distel B, Elgersma Y, Iping R. (2022) From first report to clinical trials: a bibliometric overview and visualization of the development of Angelman syndrome research. Hum Genet. Pubmed
Pandya NJ, et.al. (2022) A cross-species spatiotemporal proteomic analysis identifies UBE3A-dependent signaling pathways and targets. Mol Psychiatry. Pubmed
Duis J, et.al. (2022) A multidisciplinary approach and consensus statement to establish standards of care for Angelman syndrome. Mol Genet Genomic Med. e1843 Pubmed
Judson MC, el.al. (2021) Dual-isoform hUBE3A gene transfer improves behavioral and seizure outcomes in Angelman syndrome model mice. JCI Insight 6(20):e144712. Pubmed
Pandya NJ, et.al. (2021) Secreted retrovirus-like GAG-domain-containing protein PEG10 is regulated by UBE3A and is involved in Angelman syndrome pathophysiology. Cell Rep Med. 2(8):100360. Pubmed
Milazzo C, Mientjes EJ, et.al. (2021) Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. JCI Insight Aug 9;6(15):145991. Pubmed
Bossuyt S, et.al. (2021) Loss of nuclear UBE3A activity is the predominant cause of Angelman syndrome in individuals carrying UBE3A missense mutations. Hum Mol Genet. Pubmed
Elgersma Y & Sonzogni M. (2021) UBE3A reinstatement as a disease-modifying therapy for Angelman syndrome. Dev Med Child Neurol. Pubmed
Avagliano Trezza T, et.al. (2021) Mono-ubiquitination of Rhabphilin 3A by UBE3A serves a non-degradative function. Sci Rep. 11(1):3007. Pubmed
Den Besten I, et.al. (2020) Clinical aspects of a large group of adults with Angelman syndrome. Am J Med Genet A. Pubmed
Sonzogni M, et.al. (2020) Assessing the requirements of prenatal UBE3A expression for rescue of behavioral phenotypes in a mouse model for Angelman syndrome. Mol Autism. 11(1):70. Pubmed
Geerts-Haages A, et.al. (2020) A novel UBE3A sequence variant identified in eight related individuals with neurodevelopmental delay, results in a phenotype which does not match the clinical criteria of Angelman syndrome. Mol Genet Genomic Med. Pubmed
Zampeta IF, et.al. (2020) Conserved UBE3A subcellular distribution between human and mice is facilitated by non-homologous isoforms. Hum Mol Genet. Pubmed
Bindels-de Heus KGCB, et al. (2020) An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. 182:53–63. Pubmed
Rotaru DC et.al. (2020) Angelman Syndrome: From Mouse Models to Therapy. Neuroscience. 4522:30103-2 . Pubmed
Sonzogni M, et al. (2019) Delayed loss of UBE3A reduces the expression of Angelman syndrome-associated phenotypes. Mol Autism. Pubmed
Tonazzini I, et.al. (2019) The role of ubiquitin ligase E3A in polarized contact guidance and rescue strategies in UBE3A-deficient hippocampal neurons. Mol Autism. Pubmed
Avagliano Trezza R, et al. (2019) Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat Neurosci. 22; 1235–47. Pubmed
Wang T, et.al. (2018) Enhanced transmission at the calyx of held synapse in a mouse model for angelman syndrome. Front Cell Neurosci. Pubmed
Rotaru DC, et.al. (2018) Adult Ube3a gene reinstatement restores the electrophysiological deficits of prefrontal cortex layer 5 neurons in a mouse model of angelman syndrome. J Neurosci. 38; 8011–30. Pubmed
Sonzogni M, et.al. (2018) A behavioral test battery for mouse models of Angelman syndrome: A powerful tool for testing drugs and novel Ube3a mutants. Mol Autism. 14; 9-47. Pubmed
Judson MCC, et al. (2016) GABAergic Neuron-Specific Loss of Ube3a Causes Angelman Syndrome-Like EEG Abnormalities and Enhances Seizure Susceptibility. Neuron. 90; 56–69. Pubmed
Tonazzini I, et.al. (2016) Impaired Neurite Contact Guidance in Ubiquitin Ligase E3a (Ube3a)-Deficient Hippocampal Neurons on Nanostructured Substrates. Adv Healthc Mater 5; 850–62. Pubmed
Elgersma Y. (2015) Neurodevelopmental disease: A molecular tightrope. Nature 526; 50–1. Pubmed
Silva-Santos S, et.al. (2015) Ube3a reinstatement identifies distinct developmental windows in a murine Angelman Syndrome model. J Clin Invest. 125; 2069-76. Pubmed
Steinkellner, T. et al. (2012) Ca(2+)/calmodulin-dependent protein kinase IIα (αCaMKII) controls the activity of the dopamine transporter: implications for Angelman syndrome. J Biol Chem 287, 29627–29635. Pubmed
van Woerden, G.M. et al. (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci 10, 280–282. Pubmed
Elgersma, Y. (2007) Genetic engineering cures mice of neurological deficits: prospects for treating Angelman syndrome. Pharmacogenomics 8, 539–541. Pubmed
van den Ouweland, A.M. et al. (1999) Angelman syndrome: AS phenotype correlated with specific EEG pattern may result in a high detection rate of mutations in the UBE3A gene. J Med Genet 36, 723–724. Pubmed
Fang, P. et al. (1999) The spectrum of mutations in UBE3A causing Angelman syndrome. Hum Mol Genet 8, 129–135. Pubmed
Buiting, K. et al. (1998) Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counseling, and prenatal diagnosis. Am J Hum Genet 63, 170–180. Pubmed
Horsthemke, B. et al. (1996) Familial translocations involving 15q11-q13 can give rise to interstitial deletions causing Prader-Willi or Angelman syndrome. J Med Genet 33, 848–851. Pubmed
van den Ouweland, A.M. et al. (1995) DNA diagnosis of Prader-Willi and Angelman syndromes with the probe PW71 (D15S63). Hum Genet 95, 562–567. Pubmed
Heeft u vragen over onderzoek bij ENCORE? Of wilt u deelnemen? Neem dan contact met ons op via encore@erasmusmc.nl of angelman@erasmusmc.nl