Angelman-syndroom (AS) is een ernstige cognitieve ontwikkelingsstoornis die ongeveer 1:20.000 pasgeboren kinderen treft. AS tast vooral het centrale zenuwstelsel aan en veroorzaakt ernstige lichamelijke en leerstoornissen. Kinderen zien er bij de geboorte volkomen normaal uit, maar na het eerste levensjaar worden ontwikkelingsmijlpalen niet gehaald en stopt de ontwikkeling volledig bij een ontwikkelingsleeftijd van 2 jaar, ongeacht de werkelijke leeftijd van de patiënt. Symptomen kunnen variëren van patiënt tot patiënt, maar de meeste voorkomende omvatten ernstige verstandelijke beperking, motorische gebreken, gedragsafwijkingen, toevallen en ernstig verstoorde slaap. AS patiënten hebben een normale levensverwachting, maar hebben levenslange dagelijkse zorg nodig.
AS wordt veroorzaakt door de afwezigheid van een functioneel UBE3A eiwit. Het preklinisch onderzoek van het ENCORE expertisecentrum richt zich op het begrijpen van UBE3A functie en disfunctie, met als uiteindelijk doel om mogelijke behandelingen te ontwikkelen om AS symptomen te verlichten.
UBE3A target eiwitten identificeren.
UBE3A (oorspronkelijk geïdentificeerd als E6-geassocieerd eiwit, E6-AP) is een enzym met ubiquitine E3-ligase-activiteit. Dat betekent dat het ubiquitine moleculen aan andere eiwitten toevoegt (‘doelwit’ eiwit genoemd). Doorgaans zal het toevoegen van een ubiquitine (Ub) molecuul aan een doeleiwit de activiteit van het doeleiwit veranderen of resulteren in de afbraak van het doeleiwit. Een belangrijke lijn van onderzoek is om eiwitten die worden gemodificeerd door UBE3A te identificeren. Deze target eiwitten zullen ons belangrijke informatie verschaffen over de precieze rol van UBE3A in neuronen.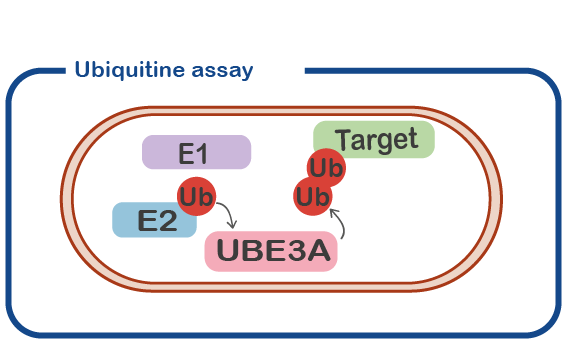
Inzicht in het effect van UBE3A mutaties.
Bij een subgroep van patiënten vinden we mutaties in het UBE3A gen die er vrij subtiel uitzien, waarbij slechts een enkel aminozuur wordt vervangen door een ander aminozuur (‘missense mutatie’ genoemd). Dit is vergelijkbaar met een spellingsfout in een woord. Wat is het effect van een dergelijke fout? Soms is het effect zeer ernstig, en hebben patiënten klassieke Angelman Syndroom kenmerken. In andere gevallen ziet de patiënt er niet uit als een typische AS patiënt, en weten we niet zeker of UBE3A daadwerkelijk niet goed functioneert of dat een ander gen gemuteerd is (zie bijvoorbeeld Geerts-Haages, Molecular Genetics and Genome Medicine, 2020). We hebben testen ontwikkeld om het effect van missense mutaties op het UBE3A-eiwit te onderzoeken, de UBE3A-ubiquitinatietest. Dit zal ons helpen bij het diagnosticeren van nieuwe patiënten, en het zal ons helpen te begrijpen hoe UBE3A functioneert.
De rol van nucleair UBE3A.
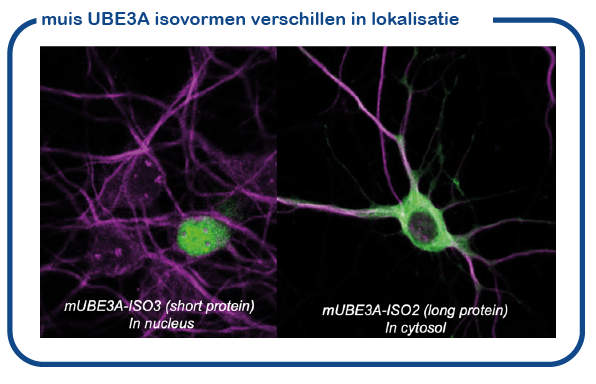
We hebben onlangs aangetoond dat de muis twee enigszins verschillende UBE3A eiwitten maakt (isoform eiwitten genoemd). Deze eiwitten verschillen een klein beetje in lengte. Het kortere UBE3A eiwit bevindt zich in de kern, de locatie waar het DNA wordt opgeslagen en uitgelezen. Het langere UBE3A eiwit is overal buiten de kern aanwezig (cytosol genoemd). We hebben onlangs ontdekt hoe dit transport naar de celkern tot stand komt (Avagliano-Trezza, Nature Neuroscience 2019). We ontdekten verder dat bij mensen en muizen het grootste deel van UBE3A zich in de kern bevindt (Zampeta, Human Molecular Genetics 2020), en we hebben aangetoond dat het nucleaire UBE3A eiwit het belangrijkst is voor de hersenfunctie. Een belangrijke onderzoekslijn van het lab is nu om de rol van UBE3A in de celkern te bestuderen.
De rol van UBE3A in de hersenen.
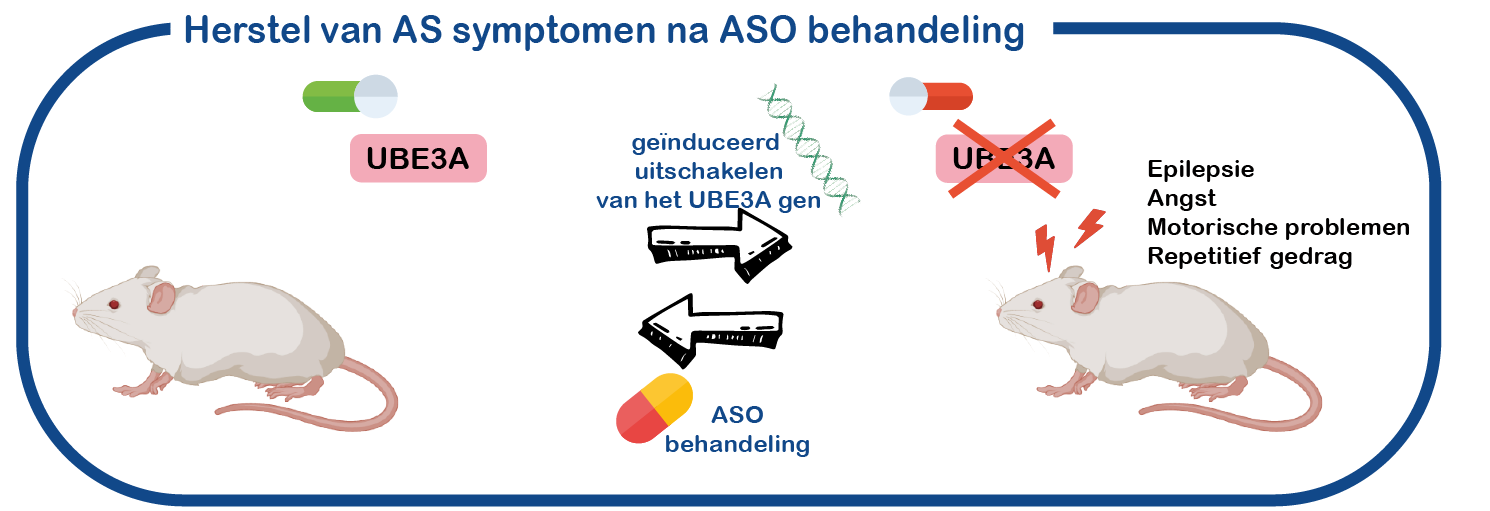
We bestuderen ook de rol van UBE3A in hersenfunctie en ontwikkeling. Omdat we dat niet in levende menselijke hersenen kunnen doen, gebruiken we hiervoor een AS muismodel dat verschillende symptomen vertoont zoals waargenomen bij AS patiënten (epilepsie, motorische gebreken, verhoogde angst, repetitief gedrag). Net als bij patiënten, mist deze muis een functioneel UBE3A gen.
Een bijkomend voordeel van ons muismodelsysteem is een ingebouwde genetische truc, waardoor we het UBE3A gen op elk gewenst moment kunnen aan- of uitschakelen. We kunnen het gen ook in- of uitschakelen in specifieke delen van de hersenen (Silva-Santos, J. Clinical Investigation, 2015; Sonzogni, Molecular Autism 2019, 2020). Dit stelt ons in staat om de rol ervan in de ontwikkeling van de hersenen te bestuderen, en ook om te bepalen welk hersengebied het meest wordt beïnvloed. Met behulp van elektrofysiologie kunnen we de geproduceerde elektrische signalen in de hersenen beoordelen en AS- en controle muizen vergelijken. Ook kunnen we ze correleren met het gedrag van deze muizen. We richten ons momenteel op de rol van UBE3A in de ontwikkeling van de hersenen en op de rol van UBE3A in het striatum, een specifiek deel van de hersenen dat mogelijk verantwoordelijk is voor motorische en gedragsproblemen.
Op zoek naar een behandeling voor AS.
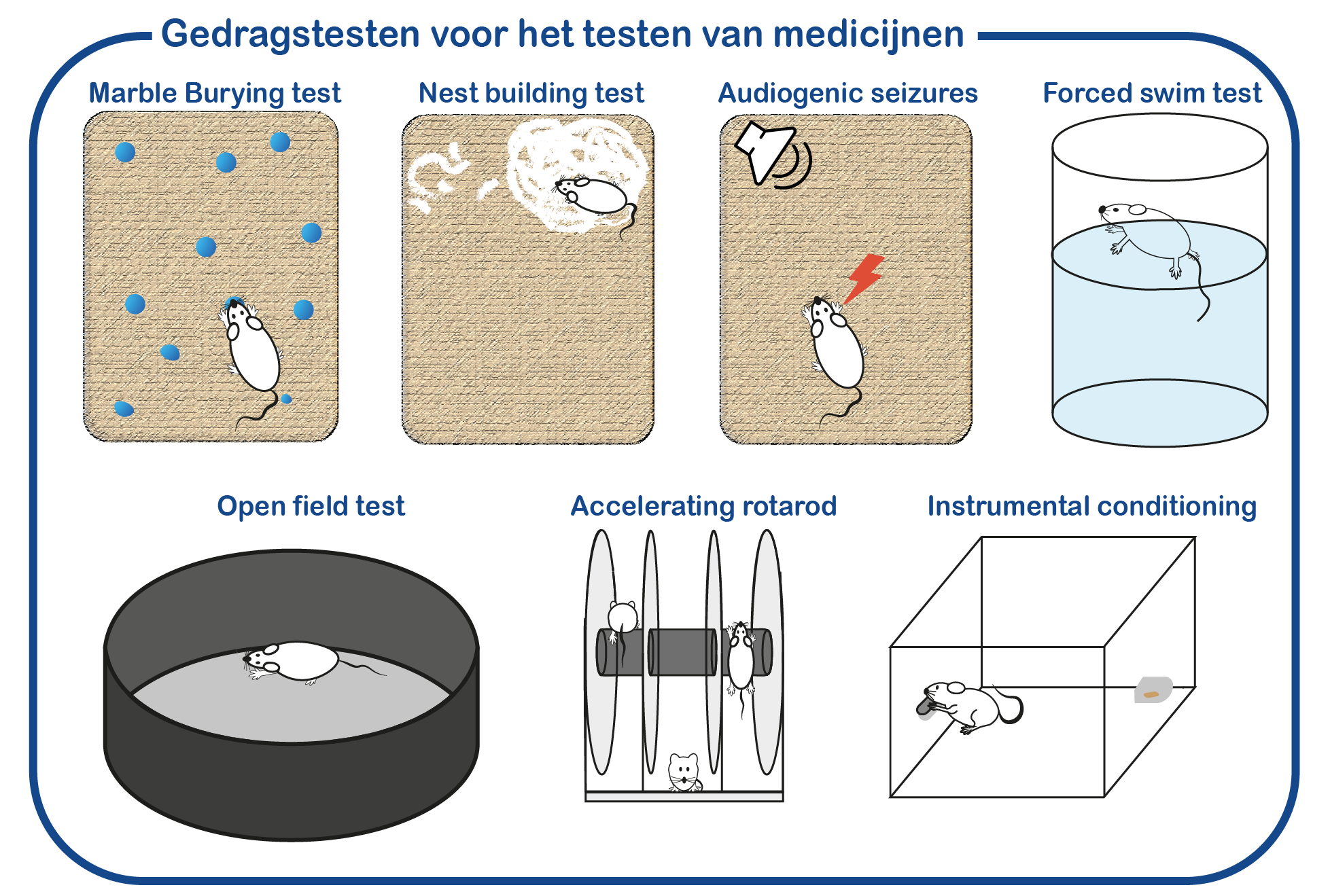
Naast fundamentele studies naar UBE3A en de rol van UBE3A in AS, onderzoeken we ook mogelijke therapeutische interventies om de symptomen van AS patiënten te verlichten. Daartoe hebben we een gestandaardiseerd screeningsprotocol ontwikkeld waarmee we het effect van medicijnen op het gedrag van AS muizen kunnen beoordelen (Sonzogni, Molecular Autism, 2018). We testen voortdurend nieuwe medicijnen die mogelijk de neurologische defecten kunnen corrigeren.
Naast conventionele therapieën, passen we ook genetische strategieën toe om UBE3A functie te herstellen. Zoals bijvoorbeeld het gebruik van antisense oligonucleotiden (ASOs of AONs genoemd) om het (vaderlijke) UBE3A gen te activeren. ASOs zijn kleine stukjes DNA/RNA die kunnen binden aan RNA moleculen. In het geval van Angelman-syndroom kunnen deze ASOs de synthese van het UBE3A eiwit herstellen door zich te richten op het Ube3a-ATS-RNA dat verantwoordelijk is voor het onderdrukken van het vaderlijke UBE3A gen. Afbraak van het Ube3a-ATS-RNA zou de UBE3A synthese bij AS patiënten herstellen.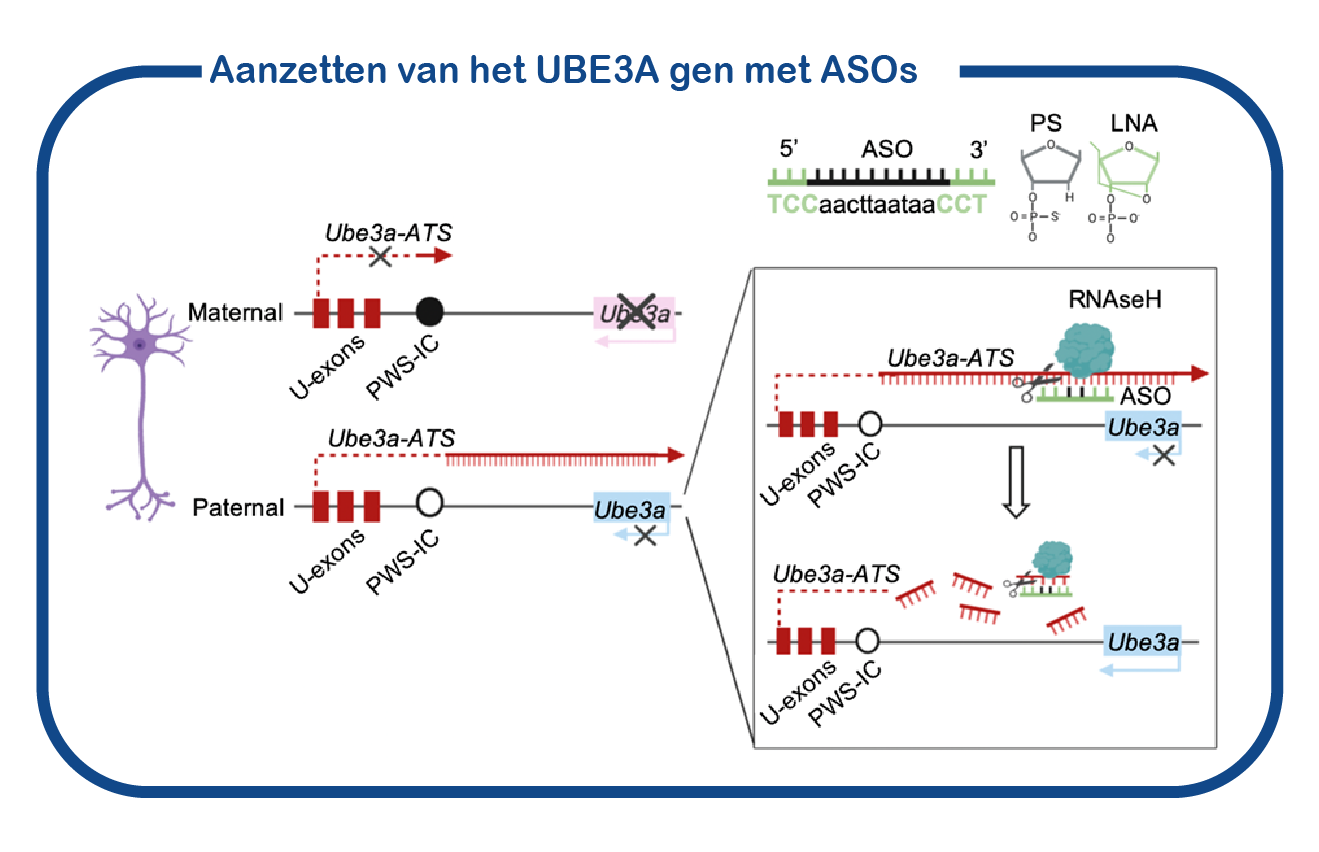
Lecoquierre F, et.al. (2024) A recurrent missense variant in the E3 ubiquitin ligase substrate recognition subunit FEM1B causes a rare syndromic neurodevelopmental disorder. Genet Med. Pubmed
Hagenaar DA, et.al. (2024) Outcome measures in Angelman syndrome. J Neurodev Disord. Pubmed
Hagenaar DA, et.al. (2023) Child characteristics associated with child quality of life and parenting stress in Angelman syndrome. J Intellect Disabil Res. Pubmed
Bindels-de Heus KGCB, et.al. (2023) Bone health in children with Angelman syndrome at the ENCORE Expertise Center. Eur J Pediatr. Pubmed
Bindels-de Heus KGCB, et.al. (2023) Hyperphagia, Growth, and Puberty in Children with Angelman Syndrome. J Clin Med. Pubmed
Keary C, et.al. (2023) Gaboxadol in angelman syndrome: A double-blind, parallel-group, randomized placebo-controlled phase 3 study. Eur J Paediatr Neurol. Aug 1;47:6-12. Pubmed
Rotaru DC, et.al. (2023). UBE3A expression during early postnatal brain development is required for proper dorsomedial striatal maturation. JCI Insight. Feb 22;8(4):e166073. Pubmed
Bindels-deHeus KGCB, et.al. (2023) Sleep problems in children with Angelman Syndrome: The effect of a behavioral intervention program. Res Dev Disabil. Feb 6;135:104444. Pubmed
Viho EMG, et.al. (2022) The Hippocampal Response to Acute Corticosterone Elevation Is Altered in a Mouse Model for Angelman Syndrome. Int J Mol Sci. Dec 24;24(1):303. Pubmed
Tanas JK, et.al. (2022) Multidimensional analysis of behavior predicts genotype with high accuracy in a mouse model of Angelman syndrome. Psychiatry. Pubmed
Lubbers K, et.al. (2022) Autism Symptoms in Children and Young Adults With Fragile X Syndrome, Angelman Syndrome, Tuberous Sclerosis Complex, and Neurofibromatosis Type 1: A Cross-Syndrome Comparison. Front Psychiatry. Pubmed
Zampeta FI, Distel B, Elgersma Y, Iping R. (2022) From first report to clinical trials: a bibliometric overview and visualization of the development of Angelman syndrome research. Hum Genet. Pubmed
Pandya NJ, et.al. (2022) A cross-species spatiotemporal proteomic analysis identifies UBE3A-dependent signaling pathways and targets. Mol Psychiatry. Pubmed
Duis J, et.al. (2022) A multidisciplinary approach and consensus statement to establish standards of care for Angelman syndrome. Mol Genet Genomic Med. e1843 Pubmed
Judson MC, el.al. (2021) Dual-isoform hUBE3A gene transfer improves behavioral and seizure outcomes in Angelman syndrome model mice. JCI Insight 6(20):e144712. Pubmed
Pandya NJ, et.al. (2021) Secreted retrovirus-like GAG-domain-containing protein PEG10 is regulated by UBE3A and is involved in Angelman syndrome pathophysiology. Cell Rep Med. 2(8):100360. Pubmed
Milazzo C, Mientjes EJ, et.al. (2021) Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. JCI Insight Aug 9;6(15):145991. Pubmed
Bossuyt S, et.al. (2021) Loss of nuclear UBE3A activity is the predominant cause of Angelman syndrome in individuals carrying UBE3A missense mutations. Hum Mol Genet. Pubmed
Elgersma Y & Sonzogni M. (2021) UBE3A reinstatement as a disease-modifying therapy for Angelman syndrome. Dev Med Child Neurol. Pubmed
Avagliano Trezza T, et.al. (2021) Mono-ubiquitination of Rhabphilin 3A by UBE3A serves a non-degradative function. Sci Rep. 11(1):3007. Pubmed
Den Besten I, et.al. (2020) Clinical aspects of a large group of adults with Angelman syndrome. Am J Med Genet A. Pubmed
Sonzogni M, et.al. (2020) Assessing the requirements of prenatal UBE3A expression for rescue of behavioral phenotypes in a mouse model for Angelman syndrome. Mol Autism. 11(1):70. Pubmed
Geerts-Haages A, et.al. (2020) A novel UBE3A sequence variant identified in eight related individuals with neurodevelopmental delay, results in a phenotype which does not match the clinical criteria of Angelman syndrome. Mol Genet Genomic Med. Pubmed
Zampeta IF, et.al. (2020) Conserved UBE3A subcellular distribution between human and mice is facilitated by non-homologous isoforms. Hum Mol Genet. Pubmed
Bindels-de Heus KGCB, et al. (2020) An overview of health issues and development in a large clinical cohort of children with Angelman syndrome. Am J Med Genet A. 182:53–63. Pubmed
Rotaru DC et.al. (2020) Angelman Syndrome: From Mouse Models to Therapy. Neuroscience. 4522:30103-2 . Pubmed
Sonzogni M, et al. (2019) Delayed loss of UBE3A reduces the expression of Angelman syndrome-associated phenotypes. Mol Autism. Pubmed
Tonazzini I, et.al. (2019) The role of ubiquitin ligase E3A in polarized contact guidance and rescue strategies in UBE3A-deficient hippocampal neurons. Mol Autism. Pubmed
Avagliano Trezza R, et al. (2019) Loss of nuclear UBE3A causes electrophysiological and behavioral deficits in mice and is associated with Angelman syndrome. Nat Neurosci. 22; 1235–47. Pubmed
Wang T, et.al. (2018) Enhanced transmission at the calyx of held synapse in a mouse model for angelman syndrome. Front Cell Neurosci. Pubmed
Rotaru DC, et.al. (2018) Adult Ube3a gene reinstatement restores the electrophysiological deficits of prefrontal cortex layer 5 neurons in a mouse model of angelman syndrome. J Neurosci. 38; 8011–30. Pubmed
Sonzogni M, et.al. (2018) A behavioral test battery for mouse models of Angelman syndrome: A powerful tool for testing drugs and novel Ube3a mutants. Mol Autism. 14; 9-47. Pubmed
Judson MCC, et al. (2016) GABAergic Neuron-Specific Loss of Ube3a Causes Angelman Syndrome-Like EEG Abnormalities and Enhances Seizure Susceptibility. Neuron. 90; 56–69. Pubmed
Tonazzini I, et.al. (2016) Impaired Neurite Contact Guidance in Ubiquitin Ligase E3a (Ube3a)-Deficient Hippocampal Neurons on Nanostructured Substrates. Adv Healthc Mater 5; 850–62. Pubmed
Elgersma Y. (2015) Neurodevelopmental disease: A molecular tightrope. Nature 526; 50–1. Pubmed
Silva-Santos S, et.al. (2015) Ube3a reinstatement identifies distinct developmental windows in a murine Angelman Syndrome model. J Clin Invest. 125; 2069-76. Pubmed
Steinkellner, T. et al. (2012) Ca(2+)/calmodulin-dependent protein kinase IIα (αCaMKII) controls the activity of the dopamine transporter: implications for Angelman syndrome. J Biol Chem 287, 29627–29635. Pubmed
van Woerden, G.M. et al. (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci 10, 280–282. Pubmed
Elgersma, Y. (2007) Genetic engineering cures mice of neurological deficits: prospects for treating Angelman syndrome. Pharmacogenomics 8, 539–541. Pubmed
van den Ouweland, A.M. et al. (1999) Angelman syndrome: AS phenotype correlated with specific EEG pattern may result in a high detection rate of mutations in the UBE3A gene. J Med Genet 36, 723–724. Pubmed
Fang, P. et al. (1999) The spectrum of mutations in UBE3A causing Angelman syndrome. Hum Mol Genet 8, 129–135. Pubmed
Buiting, K. et al. (1998) Sporadic imprinting defects in Prader-Willi syndrome and Angelman syndrome: implications for imprint-switch models, genetic counseling, and prenatal diagnosis. Am J Hum Genet 63, 170–180. Pubmed
Horsthemke, B. et al. (1996) Familial translocations involving 15q11-q13 can give rise to interstitial deletions causing Prader-Willi or Angelman syndrome. J Med Genet 33, 848–851. Pubmed
van den Ouweland, A.M. et al. (1995) DNA diagnosis of Prader-Willi and Angelman syndromes with the probe PW71 (D15S63). Hum Genet 95, 562–567. Pubmed