Tuberous sclerosis complex (TSC) is a genetic disorder affecting many different organs, including the eyes, heart, kidney, skin, lungs and the brain. While the severity of the symptoms of individual patients varies strongly, TSC symptoms associated with the brain tend to have the strongest effect on the quality of life. Most patients develop seizures, developmental delay, intellectual disability, and autism. Estimates indicate that one in 6,000 children born is affected by TSC. However, many cases remain undiagnosed as a result of mild symptoms and some TSC patients remain unnoticed until a more severely affected sibling is diagnosed with TSC. One third of patients inherit the genetic mutation from a (mildly affected) parent, while the other two thirds develop the mutation 'de novo' meaning the mutation arises spontaneously in the fetus.
TSC is caused by a mutation in either the TSC1 or the TSC2 gene, which encode the proteins hamartin and tuberin, respectively. These proteins work together to regulate the activity of the mTOR enzyme. A mutation in either gene causes the absence or malfunctioning of the respective protein. This causes mTOR to be hyperactive, which affects several crucial functions in an affected neuron.
In the lab, we are in particular interested in understanding the brain-related symptoms of TSC. In principle, we follow two main lines of research; one focusing on the causative mechanisms of TSC related epilepsy and the other on understanding why the severity of the disease is so diverse.
Epilepsy research.
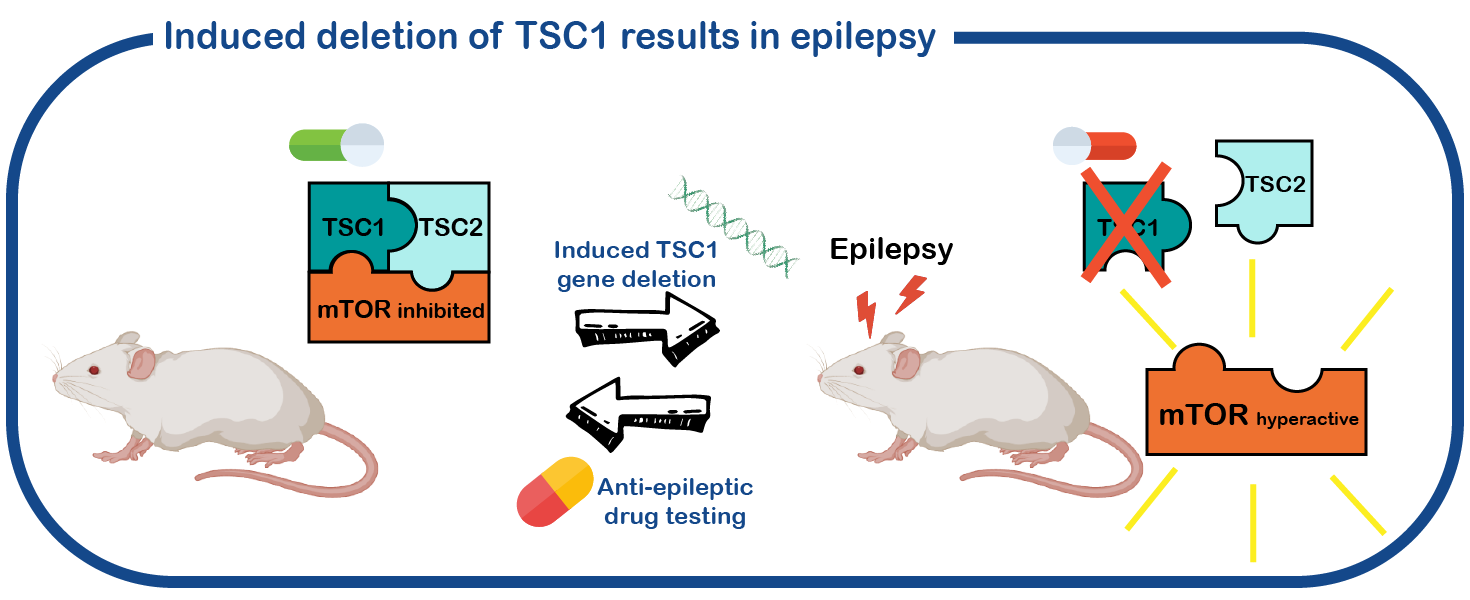
To study the epileptic brain in a physiological environment, we use a mouse-model of TSC. Our research focuses particularly on understanding how loss of a functional TSC gene causes the development of epilepsy, a process called epileptogenesis. Analyzing the electric currents in single neurons will help us to comprehend this process. The advantage of our model system lays in a genetic trick, that the TSC-causing gene TSC1 can be deleted at a desired point in time. Hence, this allows us to precisely study the cellular and molecular changes that are induced by the loss of the TSC gene, and eventually cause epilepsy. By understanding this process, we hope to develop better drugs. We also use these mice to test which anti-epileptic drugs work best for treating TSC related epilepsy (Koene, Annals of Clinical and Translational Neurology, 2019).
Discordance of TSC severity..
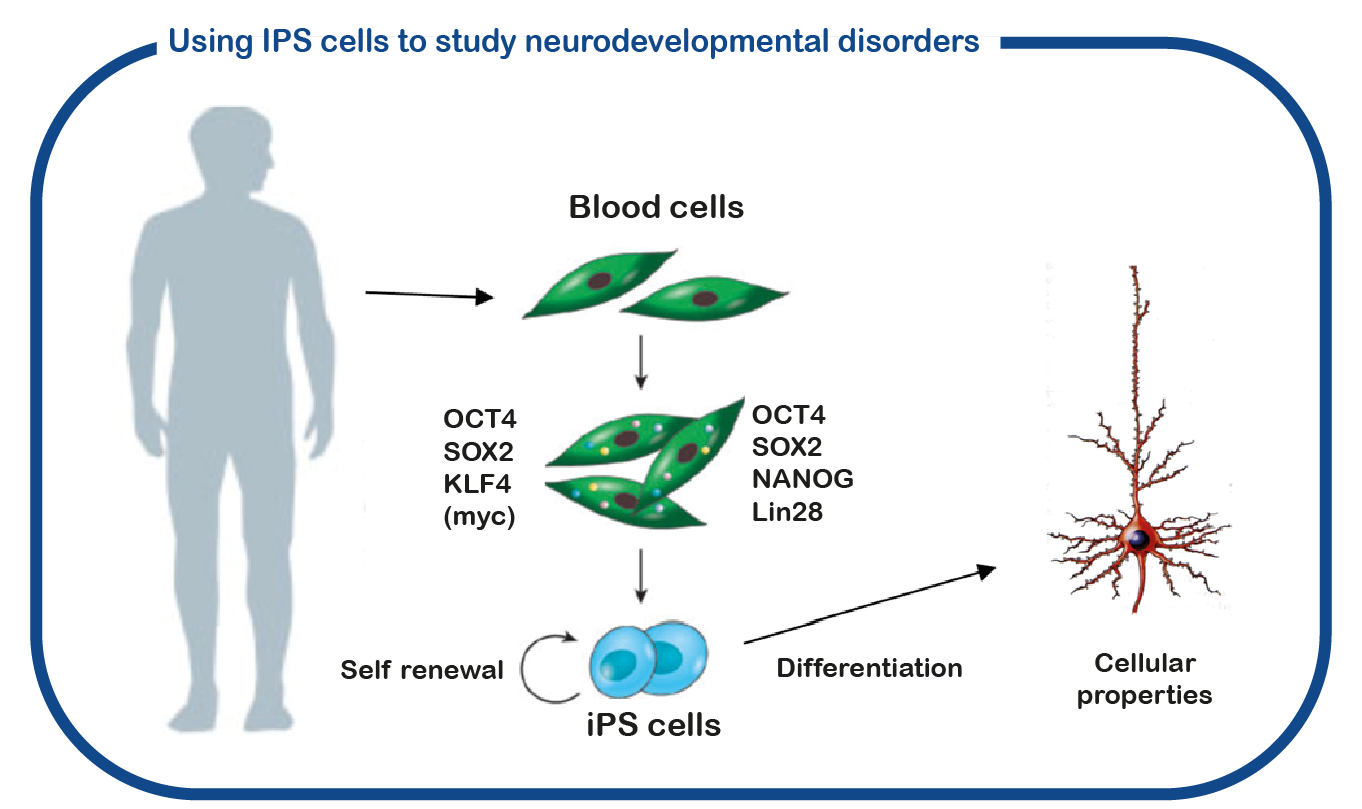
In general, TSC patients with a mutation in the TSC2 gene show more severe symptoms than those lacking TSC1. However, even when the same mutation is inherited from parent to child, their symptoms may still strongly diverge. Understanding the underlying causes for these differences may allow us to develop new treatments. To study this we cannot use mice, as they are all genetically identical and do not show such variability. Hence, we utilize induced pluripotent stem (iPS) cells. These stem cells are generated from reprogrammed blood cells donated by patients of such discordant family members. The major benefit of such iPS cells is that we can differentiate these cells into human neurons, which enables us to study human (patient) neurons in a dish. With this approach we hope to decipher factors that cause the variation in severity. These might help predict the severity of TSC for specific patients early on, enable assessment of potential treatment options, and in the long run improve disease severity and quality of life.
Heuvelmans AM, et.al. (2024) Modeling mTORopathy-related epilepsy in cultured murine hippocampal neurons using the multi-electrode array. Exp Neurol. Pubmed
Müller AR, et.al. (2024) Cannabidiol (Epidyolex®) for severe behavioral manifestations in patients with tuberous sclerosis complex, mucopolysaccharidosis type III and fragile X syndrome: protocol for a series of randomized, placebo-controlled N-of-1 trials. BMC Psychiatry. Pubmed
Müller AR, et.al. (2023) Understanding the impact of tuberous sclerosis complex: development and validation of the TSC-PROM. BMC Med. Aug 8;21(1):298. Pubmed
Lubbers K, et.al. (2022) Autism Symptoms in Children and Young Adults With Fragile X Syndrome, Angelman Syndrome, Tuberous Sclerosis Complex, and Neurofibromatosis Type 1: A Cross-Syndrome Comparison. Front Psychiatry. Pubmed
Koene LM, et.al. (2021) Identifying the temporal electrophysiological and molecular changes that contribute to TSC-associated epileptogenesis. JCI Insight. 6(23):e150120. Pubmed
Koene LMC, et al. (2019) Effects of antiepileptic drugs in a new TSC/mTOR-dependent epilepsy mouse model. Ann Clin Transl Neurol. 6; 1273–91. Pubmed
Overwater IE, et al. (2019) A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology. 93; E200–9. Pubmed
Overwater IE, et.al. (2019) Everolimus for the treatment of refractory seizures associated with tuberous sclerosis complex (TSC): Current perspectives. Ther Clin Risk Manag. 951–5. Pubmed
Mous SE, et al. (2018) Cortical dysplasia and autistic trait severity in children with Tuberous Sclerosis Complex: a clinical epidemiological study. Eur Child Adolesc Psychiatry. 27; 753–65. Pubmed
Both P, et al. (2018) Tuberous sclerosis complex: Concerns and needs of patients and parents from the transitional period to adulthood. Epilepsy Behav. Pubmed
Overwater IE, et al. (2017) Interdependence of clinical factors predicting cognition in children with tuberous sclerosis complex. J Neurol. 264; 161–7. Pubmed
Reijnders MRF, et al. (2017) Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat Commun. Pubmed
Overwater IE, et al. (2016) Genotype and brain pathology phenotype in children with tuberous sclerosis complex. Eur J Hum Genet. 24; 1688–95. Pubmed
Overwater IE, et al. (2016) Sirolimus for epilepsy in children with tuberous sclerosis complex. Neurology. 87; 1011–8. Pubmed
Overwater IE, et al. (2015) Epilepsy in children with tuberous sclerosis complex: Chance of remission and response to antiepileptic drugs. Epilepsia. 56; 1239–45. Pubmed
Goorden SMI, et.al. (2015) Intact neuronal function in Rheb1 mutant mice: implications for TORC1-based treatments. Hum Mol Genet 24; 3390–8. Pubmed
Peters JM, et al. (2014) Diffusion tensor imaging and related techniques in tuberous sclerosis complex: review and future directions. Future Neurol 8; 583–97. Pubmed
Overwater IE, et.al. (2014) Treatment of intractable epilepsy in tuberous sclerosis complex with everolimus is not yet evidence-based. Ann Neurol. Pubmed
Abs, E. et al. (2013) TORC1-dependent epilepsy caused by acute biallelic Tsc1 deletion in adult mice. Ann Neurol 74, 569–579. Pubmed
Overwater, I.E. et al. (2013) Behandelingen voor genetische neurocognitieve aandoeningen. Neuropraxis 5, 132–138. Link
van Eeghen, A.M. et al. (2013) The neuroanatomical phenotype of tuberous sclerosis complex: focus on radial migration lines. Neuroradiology 55, 1007–1014. Pubmed
Melser, S. et al. (2013) Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metabolism 17, 719–730. Pubmed
van Eeghen, A.M. et al. (2013) Central TSC2 missense mutations are associated with a reduced risk of infantile spasms. Epilepsy Res 103, 83–87. Pubmed
van Eeghen, A.M. et al. (2012) Understanding relationships between autism, intelligence, and epilepsy: a cross-disorder approach. Dev Med Child Neurol 55, 146–153. Pubmed
Hoogeveen-Westerveld, M. et al. (2012) Functional Assessment of TSC2 Variants Identified in Individuals with Tuberous Sclerosis Complex. Hum Mutat. Pubmed
van Eeghen, A.M. et al. (2012) Genotype and cognitive phenotype of patients with tuberous sclerosis complex. Eur J Hum Genet 20, 510–515. Pubmed
van Eeghen, A.M. et al. (2012) Cognitive and adaptive development of patients with tuberous sclerosis complex: A retrospective, longitudinal investigation. Epilepsy Behav 23, 10–15. Pubmed
Goorden, S.M.I. et al. (2011) Rheb is essential for murine development. Mol Cell Biol 31, 1672–1678. Pubmed
Goorden, S.M.I. and Elgersma, Y. (2011) Rheb: enrichment beyond the brain. Cell Cycle 10, 2412–2413. Pubmed
van den Ouweland, A.M.W. et al. (2011) Characterisation of TSC1 promoter deletions in tuberous sclerosis complex patients. Eur J Hum Genet 19, 157–163. Pubmed
van Eeghen, A.M. et al. (2011) Characterizing sleep disorders of adults with tuberous sclerosis complex: a questionnaire-based study and review. Epilepsy Behav 20, 68–74. Pubmed
Goorden, S.M.I. et al. (2007) Cognitive deficits in Tsc1+/- mice in the absence of cerebral lesions and seizures. Ann Neurol 62, 648–655. Pubmed
Sancak, O. et al. (2005) Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet 13, 731–741. Pubmed
Nellist, M. et al. (2005) Large deletion at the TSC1 locus in a family with tuberous sclerosis complex. Genet. Test. 9, 226–230. Pubmed
Nellist, M. et al. (2003) Regulation of tuberous sclerosis complex (TSC) function by 14-3-3 proteins. Biochem Soc Trans 31, 587–591. Pubmed
Nellist, M. et al. (2001) TSC2 missense mutations inhibit tuberin phosphorylation and prevent formation of the tuberin-hamartin complex. Hum Mol Genet 10, 2889–2898. Pubmed
Goedbloed, M.A. et al. (2001) Analysis of TSC2 stop codon variants found in tuberous sclerosis patients. Eur J Hum Genet 9, 823–828. Pubmed
Nellist, M. et al. (1999) Characterization of the cytosolic tuberin-hamartin complex. Tuberin is a cytosolic chaperone for hamartin. J Biol Chem 274, 35647–35652. Pubmed
Verhoef, S. et al. (1999) High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet 64, 1632–1637. Pubmed
van Slegtenhorst, M. et al. (1999) Mutational spectrum of the TSC1 gene in a cohort of 225 tuberous sclerosis complex patients: no evidence for genotype-phenotype correlation. J Med Genet 36, 285–289. Pubmed
Verhoef, S. et al. (1999) Malignant pancreatic tumour within the spectrum of tuberous sclerosis complex in childhood. Eur J Pediatr 158, 284–287. Pubmed
van Slegtenhorst, M. et al. (1998) Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet 7, 1053–1057. Pubmed
Wang, Q. et al. (1998) Identification of a large insertion and two novel point mutations (3671del8 and S1221X) in tuberous sclerosis complex (TSC) patients. Mutations in brief no. 119. Online. Hum Mutat 11, 331–332. Pubmed
van Slegtenhorst, M. et al. (1997) Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808. Pubmed
Rinke de Wit, T.F. et al. (1996) Expression of tyrosine kinase gene in mouse thymic stromal cells. Int Immunol. 8, 1787–1795. Pubmed
Vrtel, R. et al. (1996) Identification of a nonsense mutation at the 5′ end of the TSC2 gene in a family with a presumptive diagnosis of tuberous sclerosis complex. J Med Genet 33, 47–51. Pubmed
Halley, D.J. (1996) Tuberous sclerosis: between genetic and physical analysis. Acta Genet Med Gemellol (Roma) 45, 63–75. Pubmed
van Slegtenhorst, M. et al. (1995) Cosmid contigs from the tuberous sclerosis candidate region on chromosome 9q34. Eur J Hum Genet 3, 78–86. Pubmed
Janssen, B. et al. (1994) Refined localization of TSC1 by combined analysis of 9q34 and 16p13 data in 14 tuberous sclerosis families. Hum Genet 94, 437–440. Pubmed