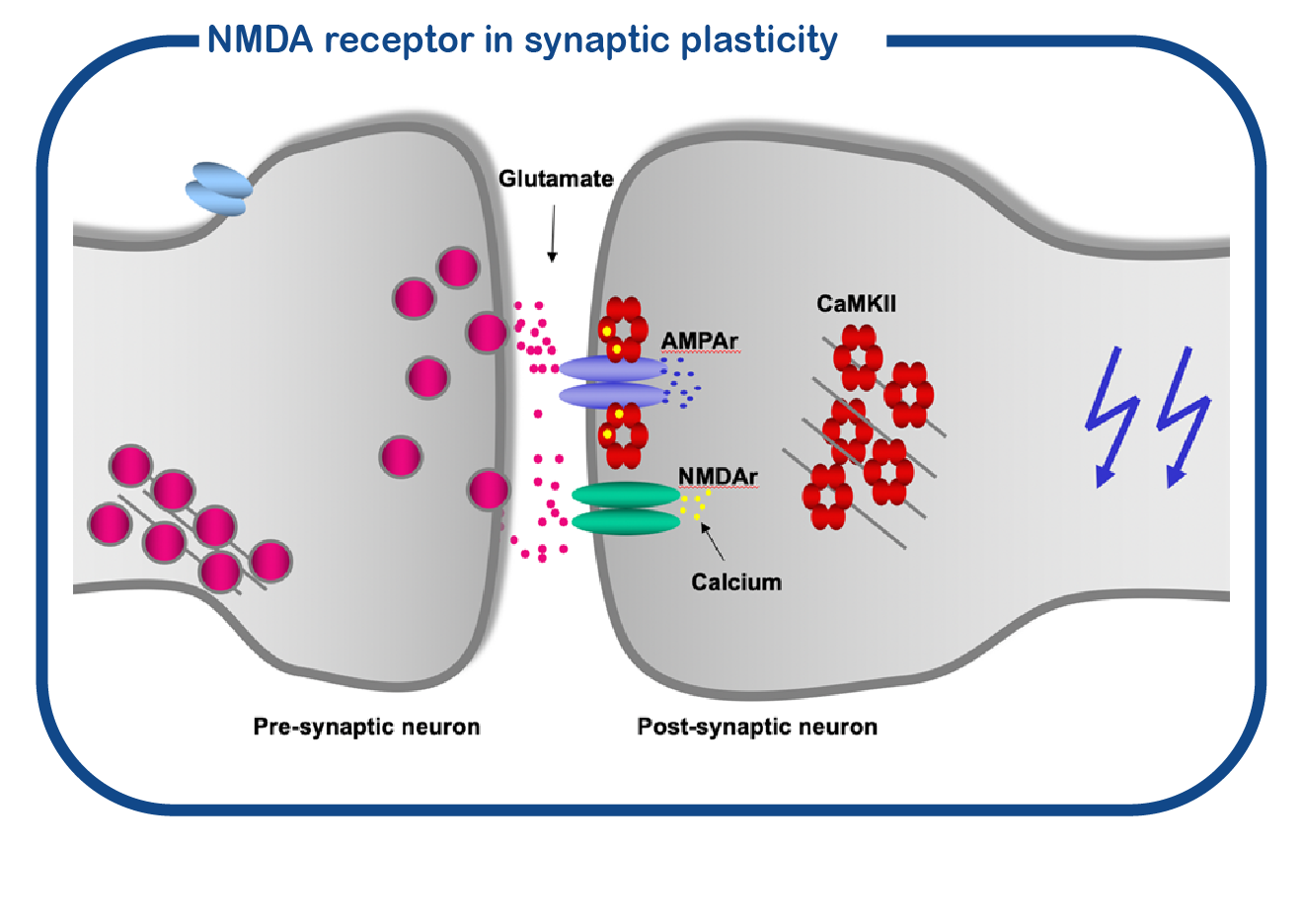
GRIN disorders are caused by mutations in the GRIN1, GRIN2A, GRIN2B, or GRIN2D genes. The GRIN proteins belong to the family of ionotropic glutamate receptors, to which also the GRIA genes belong. The GRIN proteins form a channel called NMDA receptor, the GRIA genes encode proteins for the AMPA receptor channel. The NMDA receptor is also known as ‘coincidence detector’: it will only open if the presynaptic cell releases glutamate and at the same time the postsynaptic cell fires action potentials. When this ‘coincidence’ happens, the NMDA receptor opens and allows calcium influx into the postsynaptic neuron. Calcium activates the CAMK2 enzyme, which is bound to the NMDA receptor. Activation of CAMK2 induces synaptic changes. This process is essential for learning and memory formation.
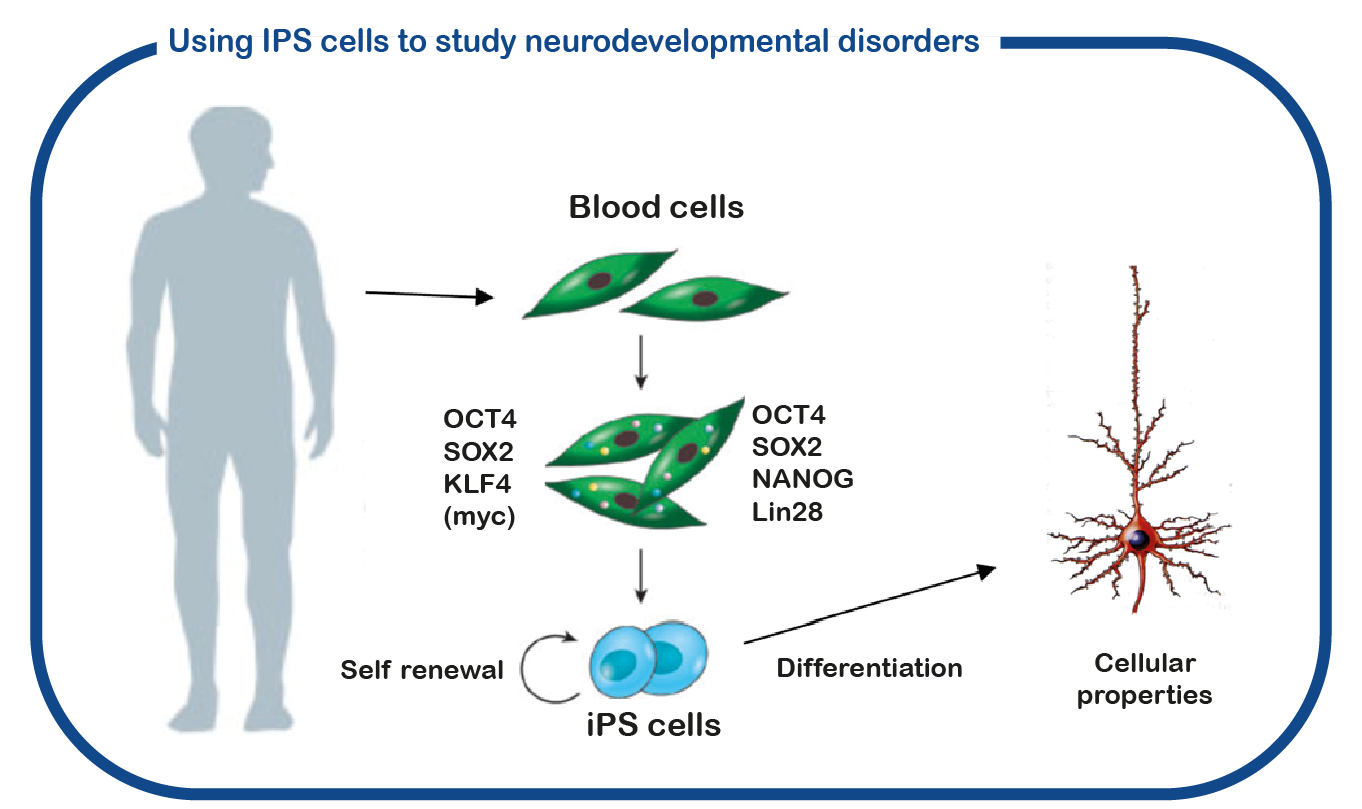
Although ENCORE has a long history of studying CAMK2 signaling (see CAMK2 page at this site), GRIN disorders are only studied recently. For that we utilize induced pluripotent stem (iPS) cells. These stem cells are generated from reprogrammed blood cells donated by patients and unaffected family members. The major benefit of such IPS cells is that we can differentiate these cells into human neurons, which enables us to study human (patient) neurons in a dish. With this approach we hope to decipher how the mutations affect GRIN function and identify novel drugs to correct these changes.